TEMA III. TUMORES DEL SISTEMA NERVIOSO CENTRAL. – I
Tema III: Temario de Neurocirugía 2020 para alumnos de Facultad de Medicina de la UAM: 2020
Facultad de Medicina de la Universidad Autónoma de Madrid.
Directores:
PARTE I.- ASPECTOS GENERALES DE LOS TUMORES CEREBRALES
1.1.- GENERALIDADES
Los tumores ocasionan el 1 % de las muertes; aunque, si se añadieran todos los tipos de metástasis, esta cifra se elevaría. Pero así como en el adulto es una enfermedad relativamente rara, en los niños es la segunda enfermedad grave más frecuente después de la leucemia.
A pesar de la importancia del órgano sobre el que asientan y al que lesionan, el tratamiento correcto puede conseguir la curación o prolongar de forma significativa la vida, aunque es fundamental un diagnóstico precoz.
Los tumores cerebrales tienen una serie de características que los diferencian del resto de tumores del organismo, como es que rara vez metastatizan fuera del cráneo y tienen una tendencia a recidivar o crecer en el mismo lugar. Otra diferencia importante es que, a medida que avanza la edad, las probabilidades de que el tumor sea benigno van en aumento; así, pasados los 70 años, los porcentajes de tumores benignos, en relación con los malignos, pueden llegar a ser igual o superior.
1.2.- FACTORES ETIOLOGICOS
A.- GENETICOS
La predisposición genética es infrecuente pero importante en las neoplasias del SNC. Hay varios hechos a favor:
1.- La existencia de una serie de enfermedades comprendidas bajo el nombre de facomatosis, que suponen una afectación simultánea neural y epitelial de extirpe congénita y con carácter hereditario (transmisión autosómica dominante). Entre ellas habría que destacar:
– Enfermedad de von Recklinghausen (Neurofibromatosis). Se asocia con la presencia de varios tipos de tumores del SNC y periférico: neurinomas o neurofibromas, gliomas o meningiomas. En el Tipo I existe una afectación del par 17 mientras que en el tipo II es característico la afectación del par 22 con presencia de neurinomas del acústico bilaterales.
– Enfermedad de von Hippel-Lindau: Se asocia con la presencia de Hemangioblastomas cerebrales o espinales. Existe una afectación del par 3. Autosómica dominante
– Enfermedad de Bourneville: Se asocia con Gliomas y hamartomas. Es una enfermedad autosómica dominante con afectación de los pares 9 y 16.
2.- Se han descrito casos de gemelos que han desarrollado el mismo tipo de tumor: meduloblastoma.
3.- Se han descrito familias con una incidencia elevada de tumores cerebrales por encima de la población normal.
Por tanto, aunque el factor genético no está claramente demostrado en la gran mayoría de los tumores, no se puede ignorar.
B.- FISICOS
1.- Traumatismos craneoencefálicos (TCE)
Incluyendo a CUSHING, se ha considerado por algunos autores la influencia de los TCE y cicatrices por fracturas con hundimientos como causa de meningiomas y gliomas.
Frente a esto hay que tener en cuenta que han aumentado los traumatismos craneales en nuestra civilización y el antecedente traumático lo presentan hasta un 35 % de personas normales.
En general no se considera el TCE como un factor etiológico, excepto en casos raros de coincidencia del tumor con cicatriz por trauma.
2.- Radiaciones
La radiación puede inducir carcinoma de piel expuesta a dosis acumulativas, pero no está demostrado que pueda provocar la aparición de tumores cerebrales.
Hay cierta evidencia de aparición de meningiomas en zonas radiadas, sobre todo en niños.
Se discute si la radioterapia podría inducir la malignización de determinados tumores, pero no se ha demostrado de forma fehaciente. Esta razón es aducida, por ejemplo, en contra de la radiocirugía, pero no se ha podido demostrar que la incidencia de los casos esporádicos radiados sea mayor que la incidencia normal.
Hoy día es motivo de preocupación la posibilidad de que las radiaciones emitidas por los teléfonos móviles pudieran inducir la generación de gliomas. Aunque aún no es posible excluir absolutamente esta posibilidad, no hay evidencia científica de que los móviles y otras radiaciones electromagnéticas puedan inducir gliomas cerebrales.
C.- QUIMICOS
1.- Derivados del antraceno.- En estudios experimentales inducen la formación del tumor tras su implante cerebral, dependiendo de la especie, edad, dosis, vía de administración, estructura química, etc. Sustancias como el benzopireno, metilcolantreno y derivados de la quinolina producirían meningiomas, fibrosarcomas, medulo
blasto
mas, gliomas y ependimomas.
2.- Nitritos y compuestos nitrosos.- Las nitrosureas son sustancias neurocarcinógenas y pueden producir tumores del SNC cuando se administran sistémicamente.
3.- Cloruro de vinilo: Su exposición ha sido implicada en la incidencia de gliomas.
D.- BIOLOGICOS
Se conoce que algunos virus, incluso obtenidos de tejidos humanos, son capaces de inducir tumores cerebrales cuando se inoculan intracerebralmente en una serie de animales. El número y tipo de tumor depende de la edad del animal y del sitio de la inyección:
Aunque aún no ha podido ser demostrada la presencia de partículas víricas en las células tumorales cerebrales del ser humano.
E.- INMUNODEPRESION
Las situaciones de inmunosupresión (síndrome de inmunodeficiencia adquirida o inmunosupresión farmacológica) pueden favorecer la aparición de tumores como los linfomas primarios cerebrales.
1.3.- PATOGENIA
En los tumores cerebrales hay que tener en cuenta una serie de formas de crecimiento que les son características, entre los que destacaremos los siguientes puntos:
1.3.1.- MECANISMOS DE CRECIMIENTO
A.- Por Expansión.- Crecen sobre un punto central produciendo compresión y destrucción del tejido adyacente.
Si la neoplasia no esta influenciada por presiones externas, suelen ser esféricas y rodeadas por una cápsula de tejido gliótico cerebral o conectivo. El crecimiento es por proliferación celular. Este tipo de crecimiento lo presentan los tumores benignos y las metástasis.

B.- Infiltración o Invasión.- Es la extensión del tumor entre los intersticios del tejido que lo rodea, pudiendo llegar a gran distancia del origen. Para que haya infiltración, las células deben tener ciertas características como capacidad de multiplicación, movilidad y poder fagocítico, posibilidad de elaboración de sustancias líticas o tóxicas, así como pérdida del control de crecimiento.
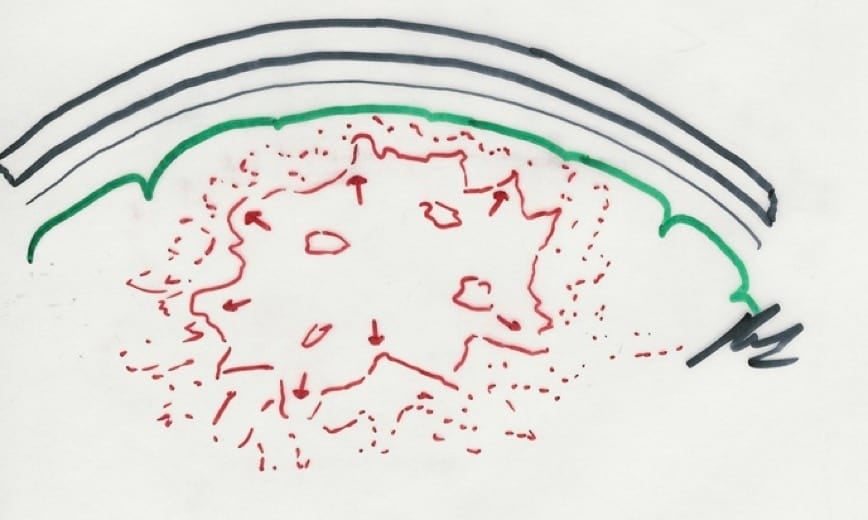
C.- Otros mecanismos.- La masa tumoral total resultante puede crecer por otros mecanismos, como el hecho de que se produzca una hemorragia intratumoral, que genere la tumoración un contenido quístico en su interior y por el edema circundante que se produce por el aumento de la permeabilidad de los vasos intra y peritumo
rales.
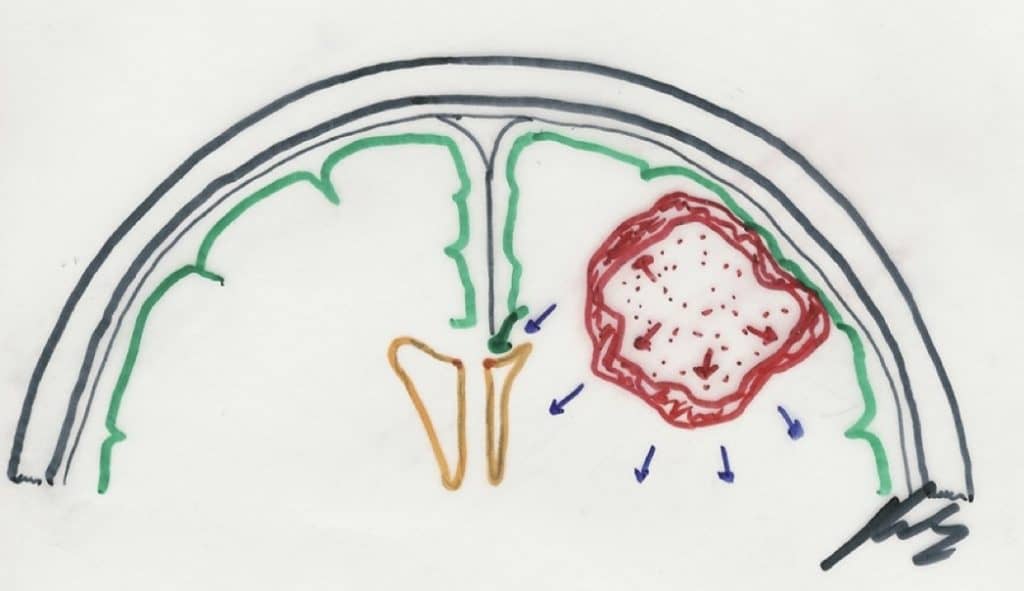
1.3.2.- FORMAS DE CRECIMIENTO
A.- Metástasis dentro del SNC:
El concepto de Metástasis implica el crecimiento tumoral en sitios lejanos, por llegada de células a través de vasos sanguíneos o linfáticos. En este sentido no es adecuado el término y es mejor utilizar el de Siembra: células en otro lugar del eje cráneo-espinal, a través del LCR.
Los tumores que presentan mayor capacidad de siembre por vía LCR son los tumores neuroectodérmicos primitivos (meduloblastoma y pinealoblastoma) (+++), los tumores de células germinales (ver tumores de la glándula pineal) (+++) y ependimoma (++).
Depende de la proximidad del foco primario al LCR, pobreza de estroma, disminución de la cohesión de las células tumora
les…, aunque están aumentando por el incremento en la supervivencia de los pacien
tes.
B.- Metástasis fuera del SNC:
De forma espontánea son excepcionales. Las localizaciones más frecuentes son en pulmón, hígado, huesos, pleura o riñón. Los tumores que pueden metastatizar son el meningioma (+++), el meduloblastoma (++) y, aún más raro, los gliomas (+).
Pero puede ocurrir por otros mecanismos yatrogénicos, por la manipulación quirúrgica y, sobre todo, tras la colocación de una derivación ventrículo-peritoneal (con siembras en peritoneo), como tratamiento de la hidrocefalia producida por un tumor con tendencia a siembras vía LCR.
C.-Crecimiento Difuso:
Originalmente un tumor es una masa visible, pero a veces las neoplasias no forman masas y se presentan de forma difusa e infiltrante en el parénquima cerebral (gliomatosis cerebri) o a nivel de todo el espacio subaracnoideo (carcinomatosis meníngea).
D.- Crecimiento Múltiple y Multicéntrico:
Se denomina múltiple cuando hay más de una masa tumoral: meningiomatosis múltiple.
El concepto de multicéntrico se reserva a la situación en que la misma masa tumoral crece en diferentes focos aparentemente independientes: gliomas multicéntricos.
E.- Recidivas:
Es la forma más frecuente de manifestarse de nuevo gran número de tumores del SNC. Aunque haya habido evidencia en las pruebas de neuroimagen de desaparición tumoral, vuelven a aparecer en el mismo lugar.
A veces se genera confusión y se denomina incorrectamente recidiva a la continuidad en el crecimiento de tumores que no han sido extirpados en su totalidad, asimilando el concepto de recidiva con el de reaparición de los síntomas.
1.3.3.- LOCALIZACIÓN
Aunque no hay una perfecta correlación con las dos formas de crecimiento anteriormente referidos, hay una clasificación general de los tumores según estén dentro del propio parénquima cerebral o se generen fuera de él, denominándose respectivamente intraparenquimatosos o extraparenquimatosos. Otra denominación similar es intraaxiales vs extraaxiales.
En cuanto a los compartimentos en los que se puede localizar un tumor, se diferencian cuatro zonas principales:
A) Supratentorial. Se afecta fundamentalmente las estructuras cerebrales corticales y subcorticales.
B) Infratentorial. Hay afectación del tronco cerebral, cerebelo y pares craneales (III en adelante).
C) Intraventricular. Cursan con hidrocefalia obstructiva.
D) Tumores de Línea Media. Es otra posible clasificación en cuanto a la localización.
Probablemente por su dificultad quirúrgica, ya que, a diferencia de las anteriores clasificaciones, en línea media puede haber una gran variedad de tumores desde el punto de vista anatomopatológico.
1.4.- EPIDEMIOLOGIA
Los tumores cerebrales presentan una incidencia aproximada en la población general de 5-10 casos nuevos /100.000 habitantes/año.
Ocasionan el 0,5 % de todas las muertes (USA, 1966) y el 2,7 % de las muertes por cáncer.
La máxima incidencia se da en varones a los 60-65 años y en mujeres a los 55-60 años. Son más infrecuentes a partir de los 65 años.
Por lo general, son más frecuentes en varones que en mujeres. Hay tumores que tienen predilección por el varón, como los tumores neuroectodérmicos primitivos (meduloblastoma o pinealoblastoma), el glioblastoma o el craneofaringioma. Otros, si embargo, tienen predilección por la mujer, entre los que destaca el meningioma.
En la edad pediátrica, son más frecuentes en la infancia y menos en la adolescencia. Las lesiones tumorales suponen un campo específico y diferencial en la infancia, con respecto al adulto, siendo, junto con las malformaciones, las entidades que más han puesto de relieve la necesidad de creación de la subespecialidad en Neurocirugía Pediátrica.
En los niños menores de 15 años el tumor cerebral es el segundo tipo más frecuente de lesión tumoral maligna después de la leucemia. Por otro lado el 15-20 % de los tumores intracra-neales ocurren en la infancia y 0,4 % de los niños hospitalizados tienen un tumor cerebral. A lo que hay que añadir que existen tumores muy específicos de la infancia (meduloblastomas, tumores de la glándula pineal, ciertos astrocitomas benignos, ependimomas, etc.) y que la frecuencia de localización tumoral supratentorial versus infratentorial se invierte en la infancia (infra > supra) con respecto a la edad adulta (supra > infra).
Como ya hemos referido, a diferencia de otros órganos en los que con la edad aumenta la malignidad, en el cerebro aumenta la benignidad con la edad. Así, en mayores de 70 años, una masa tiene un 50 % de posibilidades de ser benigna, mientras que entre los 40-50 años solo un 25-30% de probabilidad.
En cuanto a factores geográficos y sociales, las personas de raza negra parecen tener mayor frecuencia de presentación de meningiomas que los blancos y, por ejemplo, los japoneses tienen una especial mayor frecuencia de tumores de la glándula pineal.
1.5.- CLINICA
Los tumores van a dar dos tipos de síntomas:
A.- Síntomas generales.- A consecuencia de la situación de Hipertensión Intracraneal (HIC) que genera el crecimiento tumoral dentro de la estructura rígida e inextensible que es el cráneo.
El síndrome clínico de HIC consiste en cefaleas, nauseas, vómitos y edema de papila. De forma progresiva se va a ver afectado el nivel de conciencia (normal, bradipsiquia, desorientacón témporo-espacial, estupor o agitación, coma con respuesta al dolor localizándolo, flexionando extremidades o extendiendo, hasta llegar a la muerte cerebral)
Este síndrome se va a ver agudizado si se produce una hidrocefalia obstructiva o aparece un cuadro de hemorragia brusca intratumoral.
B.- Síntomas focales.- Van a ser de dos tipos:
A. Por déficit funcional, dependiendo de la localización tumoral (hemiplejia, afasia, hemianopsia, dismetría, afectación de pares craneales, etc).
B. Por irritación y exceso de función: epilepsia parcial o focal.
1.6.- DIAGNOSTICO
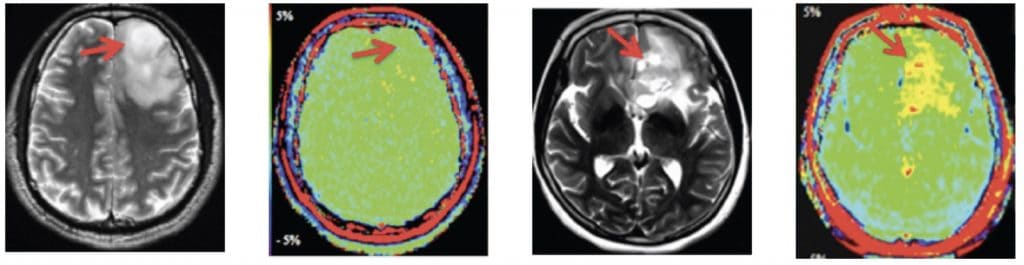
En el momento actual las pruebas diagnósticas más importantes son la Tomografia Axial Computarizada (TAC cerebral) y la Resonancia Magnética (RM). Ambas pruebas se complementan.
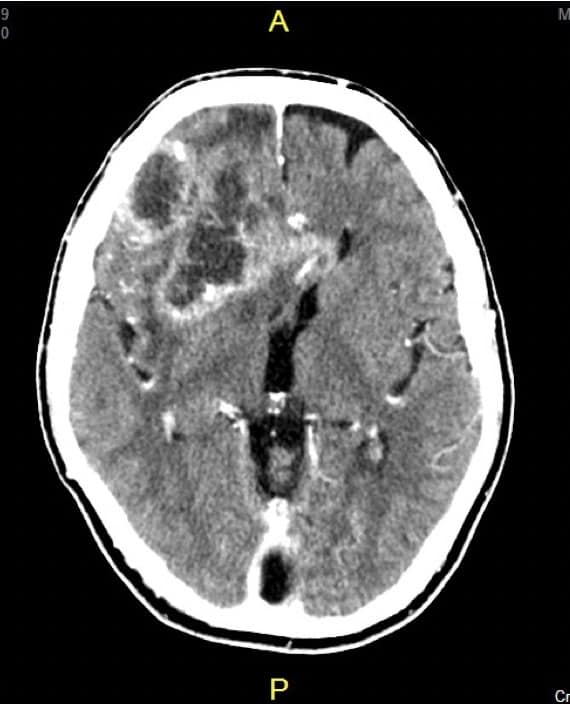
1.- TAC.- La TAC es un estudio rápido que permite ver si existen lesiones intracerebrales, lesiones calcificadas, afectación del hueso, si existen desplazamientos de estructuras… Por las características técnicas, la TAC permite visualizar muy bien las hemorragias y las calcificaciones pero, sin embargo, es una prueba muy limitada para los casos de sospecha de lesiones en la región inferior del encéfalo (cerebelo, tronco cerebral,…), debido a los artefactos técnicos que aparecen.
2.- Resonancia Magnética.- La RM aporta imágenes en los tres planos de espacio, con una visualización cada vez más perfecta de las estructuras encefálicas. Se ha convertido en el estudio fundamental para localizar y definir la lesión y sus relaciones con las estructuras adyacentes. Su inconveniente principal es que no permite ver bien el calcio y el hueso.
Ambas pruebas (TAC y RM) precisan la administración de un contraste para llevar a cabo un estudio diferencial correcto con respecto a otros tipos de lesiones del sistema nervioso.
Hoy día se complementan y han surgido técnicas de integración de las imágenes del TAC y RM en 3-D que facilitan extraordinariamente la comprensión anatómica de los tumores cerebrales.
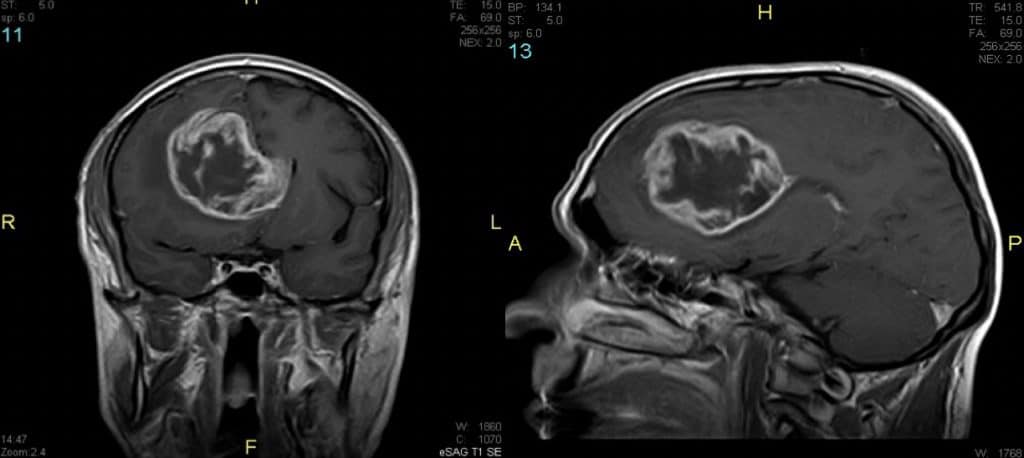
La evolución de los softwares de la RM han ofrecido técnicas como, muy recientemente, la secuencia de transferencia de protones de Amidas (3D-APT), que están permitiendo un diagnóstico muy cercano al anatomo-patológico.
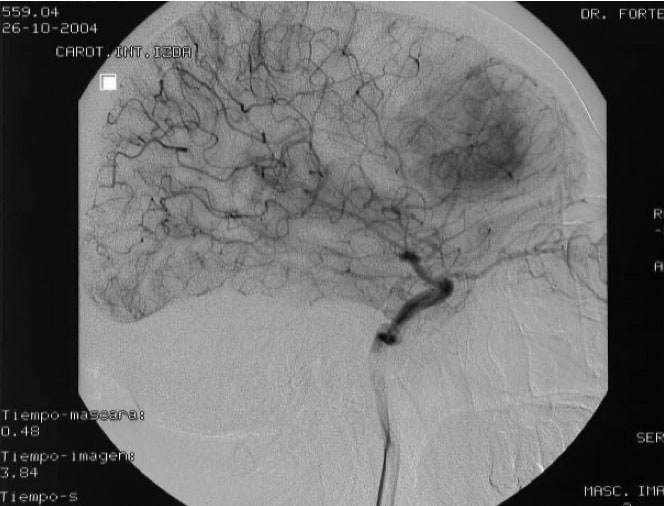
3.- Angiografía cerebral.- Es de utilidad en algunos tumores para conocer dónde se sitúan las arterias y venas y elegir la vía de abordaje más correcta a la lesión. Pero, sobre todo, es fundamental realizarla en los tumores que están muy vascularizados, como los meningiomas, para indicar a continuación su embolización, que permite cerrar los vasos que van al tumor y así facilitar la extirpación quirúrgica del tumor unos días después, al disminuir los riesgos de hemorragia.
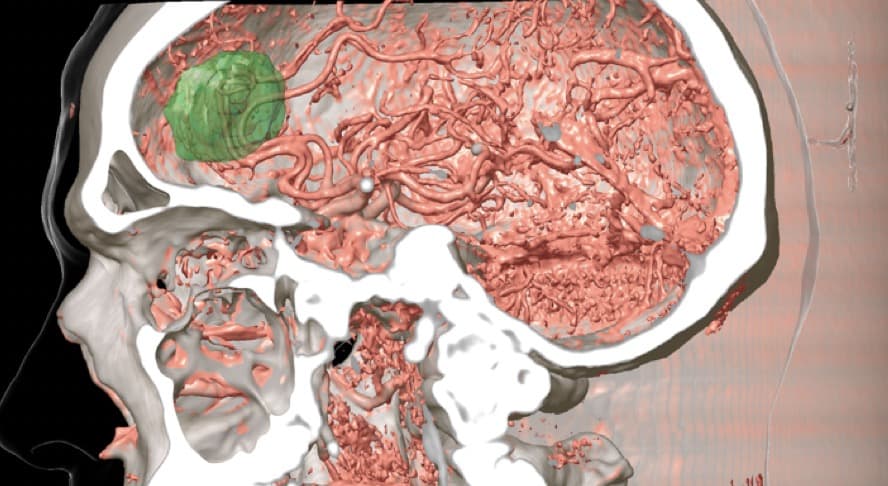
Hoy en día puede ser más útil y menos agresivo la realización de Angio-TAC cerebral para conocer la vascularización de un tumor
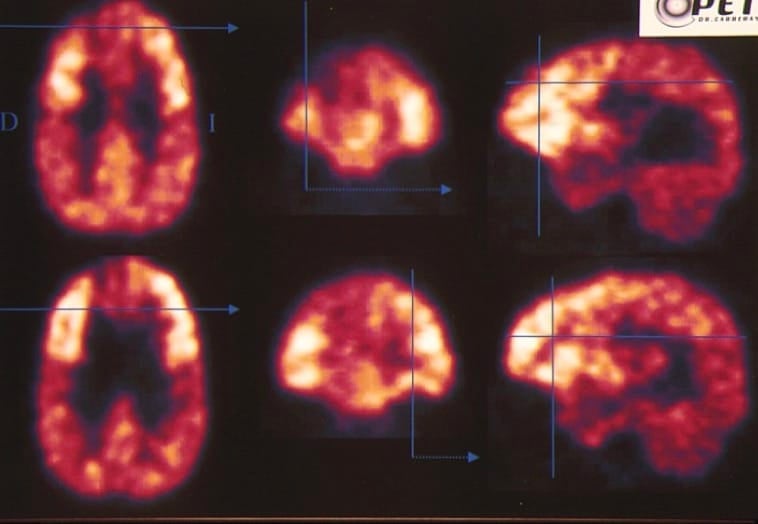
4.- Técnicas funcionales.- Las anteriores pruebas son técnicas de neuroimagen estructural o anatómica. En el momento actual se está disponiendo además de técnicas funcionales que permiten un estudio más exhaustivo de los tumores cerebrales, entre las que destacaremos:
1) Tomografía por emisión de positrones (PET).- Permite estudiar el metabolismo cerebral y diferenciar entre tumor agresivo o benigno, así como entre tumor y radionecrosis (en el seguimiento de los tumores tratados con radioterapia).
2) Espectroscopía mediante RM. – Es una técnica compleja que está facilitando, con el mismo equipo de RM y software adecuado, el “análisis bioquímico” de los componentes del tejido tumoral, diferenciando el tejido cerebral normal del anormal y, dentro de éste, el componente maligno del benigno e incluso sus posible diagnóstico histológico
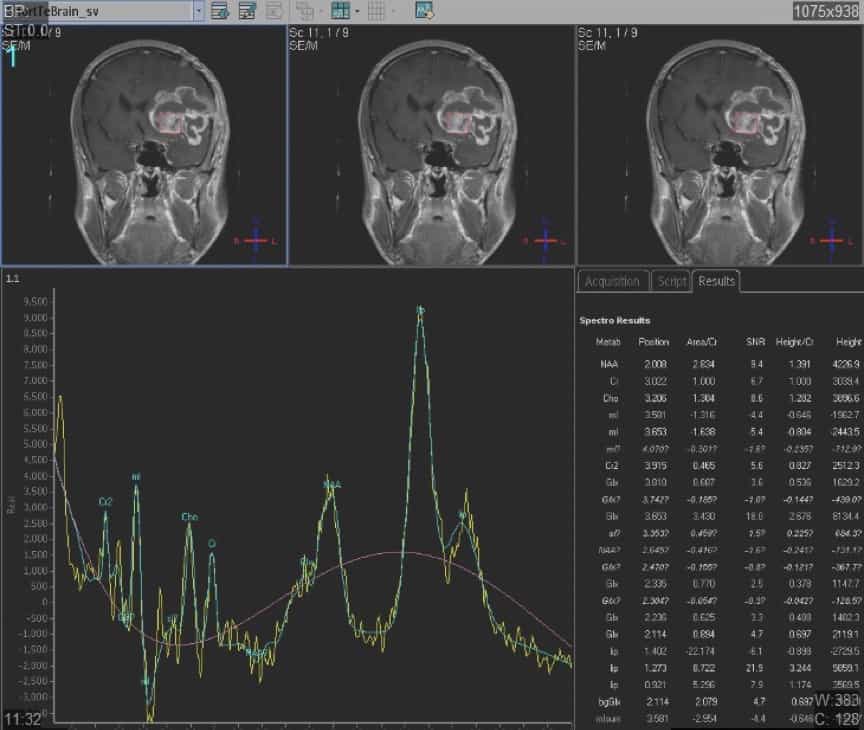
3) RM funcional.- También es una técnica muy sofisticada, pero realizada con los mismos sistemas de RM de alto campo. Con la ayuda de patrones neuropsicológicos es posible estimular y detectar la función de determinadas zonas de la corteza cerebral, anejas o próximas a la tumoración (movimiento de extremidades, sensibilidad, lenguaje, visión…). Esto ayuda sobremanera al cirujano en el diseño de la intervención quirúrgica, sobre todo en tumores que afectan la corteza cerebral, de forma que respete las zonas “no silentes”, cuya invasión produciría déficits neurológicos no deseados.
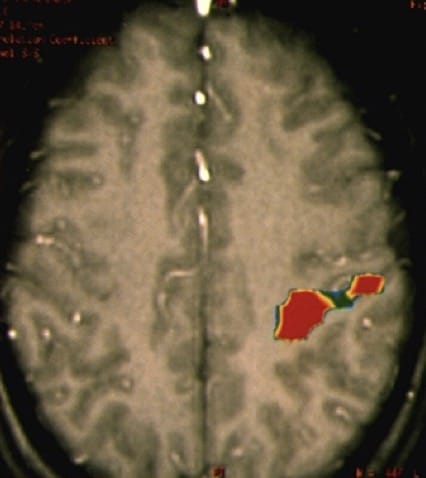
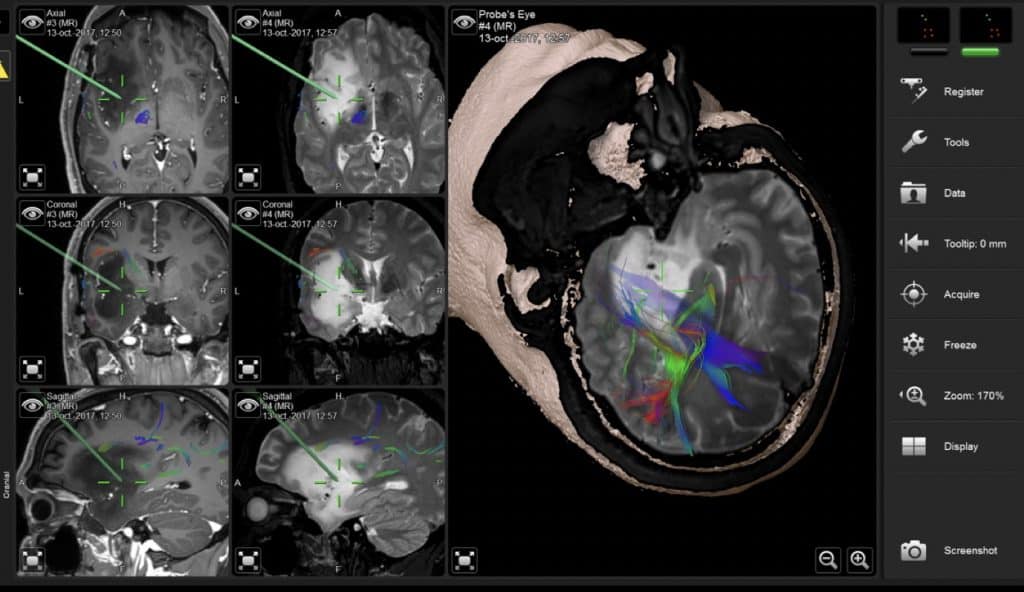
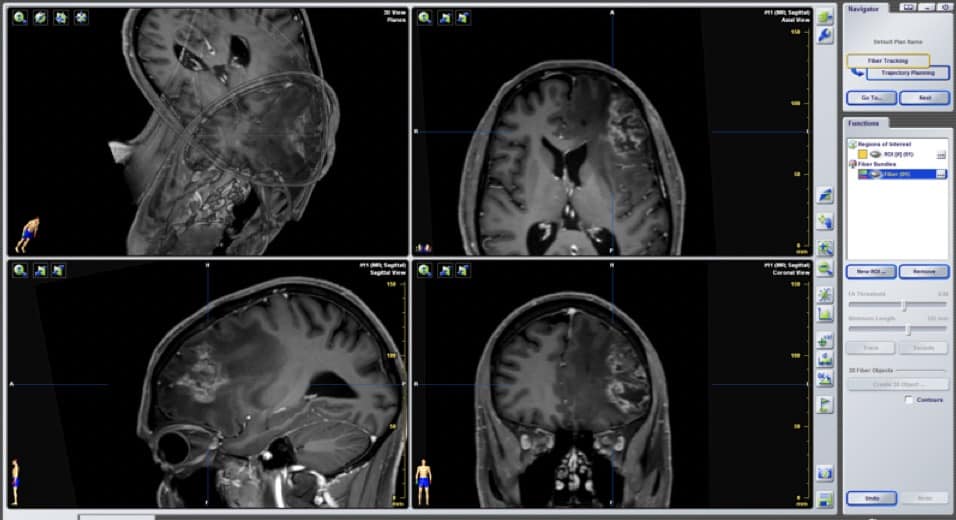
4) Tractografía.- En la RM se obtienen imágenes por tensor de difusión (DTI) que estudian los movimientos de las moléculas da agua. De esa forma se definen los tractos, haces o fascículos (vía piramidal, óptica, arcuato, uncinado…) en relación a la masa tumoral. Esto, junto con la neurofisiología y neuropsicología intraoperatoria está cambiando radicalmente el abordaje de los tumores cerebrales intraparenquimatodos, en cuanto a capacidad de exéresis radical sin lesión neurológica. A lo largo de estas lecciones veremos imágenes de DTI (Figura 17, por ejemplo).
5) Magnetoencefalografía.- Esta técnica mide los cambios de los campos magnéticos generados por la actividad eléctrica neuronal. Ayuda, por tanto, en el diagnóstico de la lesión tumoral y, acoplada a la RM, permite la localización de las zonas funcionales cerebrales, incluso con mayor precisión témporo-espacial que la RM funcional (ver Lección I).
Todas las anteriores técnicas de neuroimagen funcional tienen el enorme valor de estudiar las funciones cerebrales sin utilizar métodos invasivos. Esto hace que, aunque su coste sea alto, merezca la pena en determinados casos su utilización, para proyectar la intervención quirúrgica con menores probabilidades de producir lesiones neurológicas irreparables.
1.7.- TRATAMIENTO
El tratamiento de los tumores cerebrales es cada vez más una actividad multidisciplinaria, donde se conjugan los esfuerzos preoperatorios de los Neuroradiólogos (TAC, RM y angiografía), Neuropsicólogos (RM funcional y Magnetoencefalografía) y Especialistas en Medicina Nuclear (PET), para realizar un correcto estudio.
Durante la intervención quirúrgica los Neurofisiólogos (registro EEG de corteza cerebral, Estimulación cortical, Potenciales evocados…), Neuropsicólogos, junto a los Neurocirujanos y Neuroanestesistas, para conocer y respetar durante la resección quirúrgica las zonas funcionales importantes.
Y los Oncólogos Médicos y Radioterapeutas para completar el tratamiento tras la intervención quirúrgica.
Los neurocirujanos están continuamente sofisticando sus técnicas quirúrgicas. En primer lugar, obteniendo en los estudios preoperatorios una buena visión del tumor y zonas adyacentes, funcionalmente importantes o no, incluso con imágenes 3D y programas que simulan la vía de abordaje.
Con este objetivo se han diseñado complejos sistemas de neuroimagen quirúrgica (Neuronavegadores), que además guían al cirujano durante su resección quirúrgica, para garantizar que sea completa y respetuosa con los márgenes no tumorales. En el momento actual, prácticamente todos los neurocirujanos utilizan técnicas microquirúrgicas, ayudados de microscopios muy sofisticados, que le permiten moverse desde muy diferentes ángulos con una excelente visión en cuanto a luz y zoom variable del campo operatorio.
Con el fin de respetar el tejido noble y sólo resecar el tumoral, hay multitud de técnicas y equipamientos. Son de destacar los Láser de CO2 y Nd-YAG (hoy día en desuso) y el aspirador ultrasónico. Este último equipo permite a la vez la emisión de ultrasonidos que destruyen milimétricamente el tejido sin tocarlo, formándose una ultrafragmentación en partículas que son aspiradas de forma continua, todo ello bajo irrigación de suero salino.
Aunque es obvio que el mejor neurocirujano no necesariamente es el que más equipamiento tiene a su alrededor, sino el que se conoce mejor la anatomía y función de la zona a intervenir, sabe guiarse por las rutas anatómicas (diferentes en cada paciente) y respetar con su técnica cuidadosa las zonas nobles, aplicando en el momento preciso las tecnologías sofisticadas que complementan su capacidad quirúrgica personal.
Tras la intervención quirúrgica, dependiendo del tipo de lesión extirpada o biopsiada, entrarán en juego, si está indicado, los especialistas en radioterapia y quimioterapia. En cuanto a la Oncología Radioterápica, se ha pasado de la radioterapia convencional a técnicas muy sofisticadas de irradiación más local y precisa (Radioterapia Estereotáxica Fraccionada) o incluso a la capacidad de hacer converger numerosos haces de radiación para producir una necrosis en una zona pequeña, lo que permite tratar lesiones benignas (Radiocirugía).
Iguales avances se están produciendo en Oncología Médica, con nuevos fármacos y pautas de quimioterapia que sí consiguen pasar la barrera hematoencefálica y, por tanto, ser más activos en los tumores cerebrales agresivos.
PARTE II.- CLASIFICACIÓN DE LOSTUMORES CEREBRALES
A continuación se presentan una serie de cuadros referentes a la clasificación anatomopatológica y de frecuencia con respecto a la edad y localización. Se aconseja que estas clasificaciones sean revisadas una vez estudiados los temas siguientes. De forma que sirvan de repaso y de esquema para tener una visión global de los principales tipos de tumores.
2.1.- CLASIFICACION ANATOMOPATOLÓGICA
En 1979 la OMS realizó una clasificación de los tumores cerebrales basado en la célula de origen y estratificando los tumores en grados según su agresividad:
Grado I: Tumores circunscritos, de lento crecimiento y bajo potencial de conversión a un tumor de mayor malignidad.
Grado II: Tumores de bordes difuso, lento crecimiento y algunos con tendencia a progresar a tumores de mayor complejidad
Grado III: Tumores infiltrantes con células atípicas o anaplásicas y mayor número de mitosis
Grado IV: Tumores de rápido crecimiento con alta tasa mitótica, pudiendo presentar vasos de neoformación y áreas de necrosis.
La investigación en los marcadores moleculares de tumores y su valor pronóstico han contribuido a la última clasificación de la OMS de 2016, siendo los tumores de estirpe glial los que más modificaciones han tenido dentro de la clasificación:


2.2.- CLASIFICACION POR FRECUENCIA SEGUN LA EDAD
La incidencia de los tumores es diferente según la edad, por lo que consideramos también útil esta clasificación:
1. NIÑOS
INFRATENTORIALES (60%)
– Astrocitomas de bajo grado:
o Tronco cerebral
o Cerebelo
– Meduloblastomas
– Ependimomas del IV ventrículo
– Quistes Dermoides/Epidermoides
SUPRATENTORIALES (40%)
– Craneofaringiomas
– Tumores de la Región Pineal
– Astrocitomas (Mayor proporción de bajo grado que en el Adulto)
2. ADULTOS
SUPRATENTORIALES (80-85%)
Astrocitomas de alto grado
Meningiomas
Metástasis
Adenomas de Hipófisis
Oligodendrogliomas
Craneofaringiomas
INFRATENTORIALES (15-20%)
Neurinoma
Metastasis
Meningioma
Hemangioblastoma
Quistes Dermoide/Epidermoide
2.3.- CLASIFICACION POR FRECUENCIA SEGUN LA LOCALIZACION
Según la localización de los tumores también tienen una incidencia diferente:
2.3.1.- SUPRATENTORIALES
A.- Hemisféricos
a) Intraparenquimatosos
1.- Astrocitomas: alto y bajo grado
2.- Oligodendrogliomas
3.- Metástasis
4.- Linfoma
5.- Otros
b) Extraparenquimatosos
1.- Meningiomas
2.- Dermoide/Epidermoide
B.- Region Selar/Paraselar
1.- Adenoma de Hipófisis
2.- Craneofaringioma
3.- Glioma del Nervio Optico
4.- Meningioma
5.- Dermoides/Epidermoides
C.- Region Pineal
1.- Tumores de células germinales
2.- Tumores no germinomatosos
3.- Teratomas
4.- De células pineales
2.3.2.- INTRAVENTRICULARES
1.- Ependimoma
2.- Papiloma/Carcinoma de Plexos Coroides
3.- Quiste Coloide del III Ventrículo
4.- Meningioma
5.- Otros: Germinoma, Teratoma, Craneofaringioma
2.3.3.- INFRATENTORIALES o de FOSA POSTERIOR
A.- Intraparenquimatosos
1.- Metástasis
2.- Hemangioblastoma
3.- Meduloblastoma
4.- Astrocitoma de Cerebelo
5.- Astrocitoma de Tronco Cerebral
B.- Extraparenquimatosos
1.- Neurinoma/Neurofibroma
2.- Meningioma
3.- Quiste Dermoide/Epidermoide
2.4.- TUMORES DE LINEA MEDIA
Es otra forma de clasificación en cuanto a localización, aunque menos usada. En general son más frecuentes en niños.
Los más frecuentes, de delante a atrás:
1.- Tumores de la Region Selar/Paraselar
1.- Adenomas de Hipófisis (Adultos)
2.- Craneofaringiomas
3.- Glioma del Nervio Optico
4.- Meningioma del Tubérculo de la Silla Turca (Adultos)
5.- Quistes Dermoides/Epidermoides
2.- Tumores del III Ventriculo
1.- Quiste Coloide
3.- Tumores de la Region Pineal
4.- Tumores del IV Ventrículo
1.- Ependimoma
2.- Papiloma de Plexos Coroides (Adultos jóvenes)
5.- Tumores del Tronco Cerebral
1.- Astrocitomas (Bajo Grado)
6.- Tumores del Vermis Cerebeloso
1.- Meduloblastoma
2.- Astrocitoma (También en Hemisferios Cerebelosos)
PARTE III.- DESCRIPCION DE LOS TIPOS DE TUMORES CEREBRALES
A continuación realizaremos una descripción de las características clínicas, radiológicas y de tratamiento de cada tipo de tumor.
Se va a seguir una clasificación acorde con el posible origen celular de cada tumor, siguiendo en gran parte la nueva clasificación de la OMS.
En este sentido, de forma práctica y didáctica, podemos diferenciar varios tipos de células que implican al Sistema Nervioso, de las que podrían originarse los tumores:
1. Neuronas
2. Glía
3. Meninges
4. Vainas nerviosas
5. Glándulas: Hipófisis y Pineal
6. Plexos coroides
7. Vasos
8. Restos embrionarios
9. Hueso
10. Metástasis
3.1.- TUMORES DE EXTIRPE NEURONAL
3.1.1.- TUMOR NEUROEPITELIAL DISEMBRIOPLÁSICO (DNT)
Tumor descrito en los años 80, formado fundamentalmente por neuronas y que se encuentra prácticamente de forma exclusiva en los pacientes con epilepsia fármaco-resistente que son intervenidos quirúrgicamente para controlar sus crisis.
Son benignos, intraparenquimatosos, de pequeño tamaño y se localizan en corteza cerebral. Son muy raros. La clínica es, como ya hemos referido, de epilepsia resistente a la medicación.
El diagnóstico se lleva a cabo tras realizar la RM en el protocolo de estudio de este tipo de pacientes. Suelen ser hipodensos, apenas captan contraste y pueden tener componentes quísticos. El diagnóstico definitivo se realiza tras técnicas específicas de estudio anatomopatológico.
La resección quirúrgica se hace siguiendo las metodologías propias de una Unidad de Cirugía de la Epilepsia. La finalidad es extirpar una lesión tumoral y erradicar las crisis.
Es excepcional que recidiven.
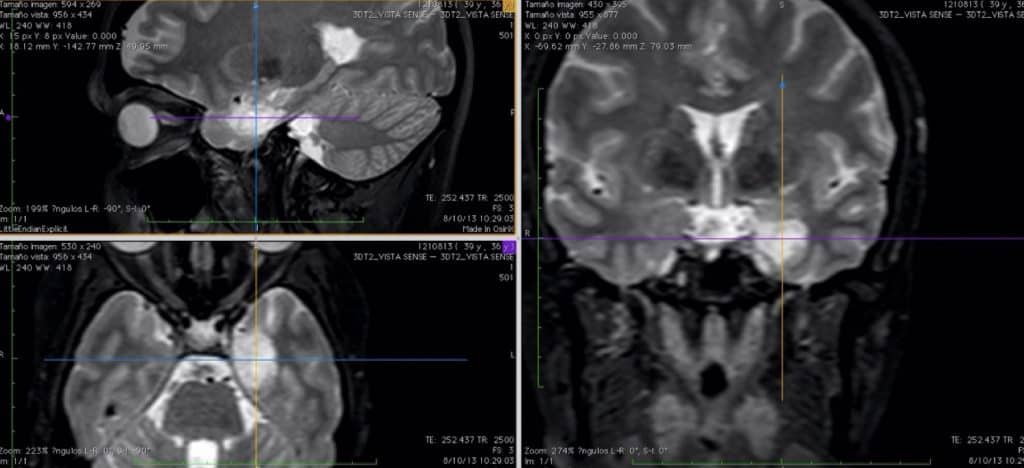
3.1.2.- GANGLIOCITOMAS y GANGLIOGLIOMAS
Tumores formados por mezcla de células neuronales y gliales maduras. El componente glial es el que marca el grado de agresividad. En el gangliocitoma hay predominio de neuronas y en el ganglioglioma predominan las células gliales.
Son poco frecuentes (0’4-1’7%) y aparecen en niños y adultos jóvenes. Suelen tener calcificaciones y una localización preferente en el lóbulo temporal, donde originan crisis epilépticas rebeldes a tratamiento médico como síntoma fundamental.
El diagnóstico se realiza con RM. Suelen tener características similares a los astrocitomas benignos, con calcio en su interior y débil captación de contraste. Rara vez tienen edema peritumoral.
El tratamiento quirúrgico se ha de llevar a cabo siguiendo los patrones referidos en el DNT. En caso de recidiva o de que existan patrones de agresividad histológicos, hay que plantearse completar el tratamiento con radioterapia.
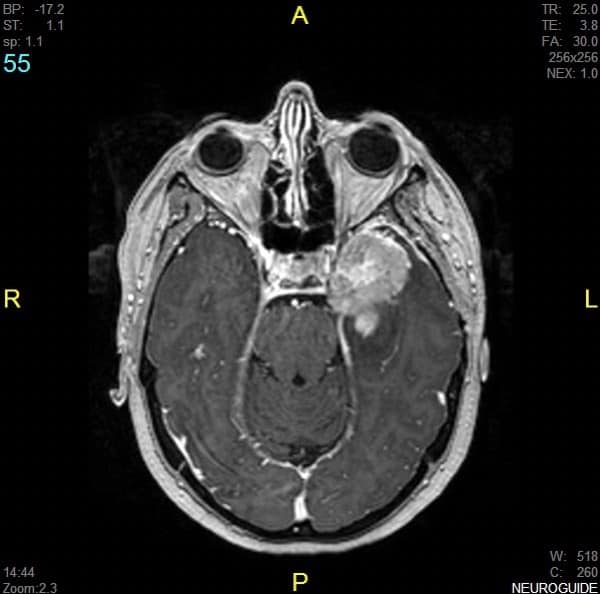
3.1.3.- MEDULOBLASTOMA
Pertenece al grupo de los llamados tumores neuroectodérmicos primitivos (PNET). Es un tumor embrionario pobremente diferenciado, de gran agresividad. Suele localizarse en vermis cerebeloso y presentarse en edad juvenil e infantil.
Es el tumor pediátrico más frecuente (20-35 % de los tumores infantiles del SNC) y supone el 4-10 % de las series tumorales.
Es mas frecuente en sexo masculino. Más del 60% aparece antes de los 14 años, con una máxima incidencia entre los 8 y 12 años. Aunque tiene otro pico de incidencia, menos acusado, entre los 20 y 25.
Se localiza preferentemente en la fosa posterior, con mayor frecuencia en el vermis cerebeloso e invade el techo del IV ventrículo.
3.1.3.1.- CLINICA
Produce fundamentalmente un síndrome cerebeloso de línea media (ataxia de la marcha, dismetría…) y un síndrome de hipertensión intracraneal (HIC) por la hidrocefalia que produce al obstruir el IV ventrículo (cefaleas, vómitos, disminución de visión por el edema de papila…).
El meduloblastoma tiene una gran tendencia a generar siembras en el eje cráneo-espinal, a través del LCR.
3.1.3.2.- DIAGNOSTICO
La Rx de cráneo puede dar signos de HIC crónica (reapertura de suturas, huellas digitiformes…). La TAC muestra una masa tumoral en fosa posterior, de límites relativamente homogéneos, que capta contraste, con zonas hipodensas en su interior (necrosis) y edema circundante.
La RM complementa al TAC, definiendo aún mejor los límites anatómicos.
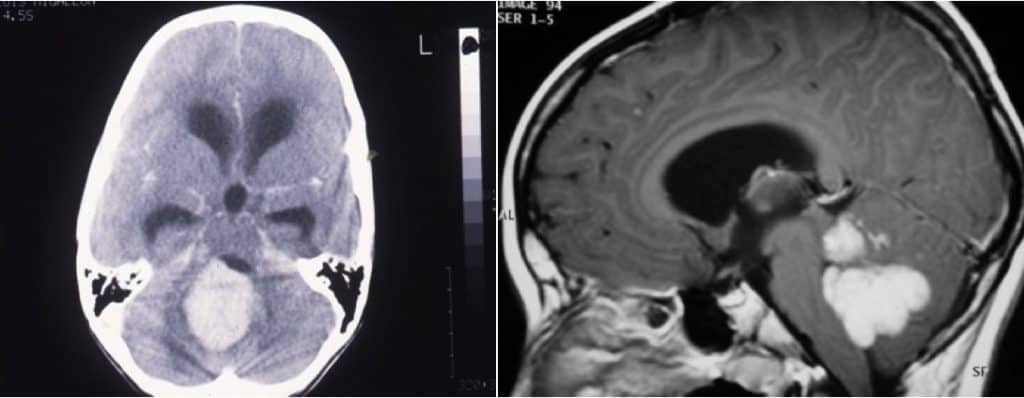
La angiografía puede ser útil, para definir la vascularización tumoral
Ha de completarse el manejo diagnóstico analizando el LCR. Durante el seguimiento han de realizarse estudios de RM de todo el neuroeje, para detectar posibles siembras.
3.1.3.3.- TRATAMIENTO
El tratamiento es complejo. Hay que programar la intervención quirúrgica con la finalidad de realizar una extirpación lo más completa posible.
A partir de aquí, en conjunción con Oncología Médica y Radioterápica se ha de generar un programa de actuación en el que se incluye la radiación de la fosa posterior, radioterapia de todo el neuroeje y quimioterapia (general e intratecal). Todo ello dependiendo de cada caso, edad, su evolución y si se trata de la primera actuación o de una recidiva.
3.1.3.4.- PRONOSTICO
Con las actuales pautas de tratamiento, este tumor tan altamente agresivo se controla relativamente, con una supervivencia del 50 % a los 3 años, 30 % a los 5 años y 20 % pasados los 10 años.
3.2.- TUMORES DE EXTIRPE GIAL
Las células gliales se dividen en astrocitos, oligodendrocitos y células de microglía. Como células especiales que revisten el sistema ventricular, los ependimocitos. Cada uno de estos tipos celulares pueden dar lugar a tumores. (astrocitomas, oligodendrogliomas, ependimomas) . Los tumores derivados de la microglía se identifican con los linfomas primarios cerebrales. Los gliomas constituyen la mayor parte de los tumores del SNC. El subgrupo más frecuente e importante es el de los Astrocitomas.
Con la nueva clasificación de la OMS para los tumores gliales es preciso, no sólo realizar una biopsia del tejido, sino que se precisa la determinación de marcadores tumorales inmuhohistoquímicos propios de los gliomas. Son muchos los marcadores que se están describiendo según los tipos de tumores gliales pero los más importantes por su implicación pronostican y de tratamiento son los siguientes:
Codelección de los cromosomas 1p/19q característica de los oligodendrogliomas. Se asocia con un curso mas lento de la enfermedad y mayor sensibilidad a los tratamientos de radio y quimioterapia.
Mutación del gen IDH1 e IDH2 (mutación isocitrato deshidrogenasa). Su presencia en los gliomas indica un mejor pronóstico que si no existe esta mutación
Metilación del gen MGMT (metilación del promotor de O6-methylguanine-DNA methytransferasa). Su presencia indica que es más favorable al tratamiento con agentes alquilantes como la Temozolamida
Amplificacion del gen EGFR
Mutacion P53
3.2.1.- ASTROCITOMA
El astrocitoma es un tumor descrito por Virchow en 1860. Se han establecido varios subtipos dependiendo de la implicación además de los oligodendrocitos y ependimocitos y del tipo de crecimiento. Para mayor sencillez y claridad es preferible mantener la clasificación de Kernohan (1949) en grados del I al IV dependiendo de la menor o mayor agresividad o malignidad.
El Glioblastoma es un tumor muy agresivo. En los últimos años, la realización de los estudios inmunohistoquimicos han contribuido a modificar la clasificación en distintas variedades con diferentes respuestas a los tratamientos
3.2.1.1.- INCIDENCIA
El astrocitoma supone el 11-13 % de todos los tumores cerebrales primarios, siendo el segundo en frecuencia después del glioblastoma (si aceptamos esta diferencia). Puede llegar a ser el 25-30 % de todos los gliomas.
Aparecen en personas de edad media de la vida o en la juventud, con un pico de edad a los 35 años y son algo más frecuentes en el varón. Los astrocitomas benignos (Grado I-II) predominan en las décadas de los 30-40 años, mientras que los más agresivos (Grado III-IV) se presentan a los 40-50 años.
3.2.1.2.- LOCALIZACION
En los adultos, la frecuencia en la localización anatómica podríamos asimilarla al porcentaje de volumen de la zona encefálica en la que se pueden presentar. Así, es más frecuente en el lóbulo frontal, en relación a los otros lóbulos cerebrales. La frecuencia de aparición en zonas subcorticales o talámicas es menor, siendo más en cerebelo y excepcionales en tronco cerebral.
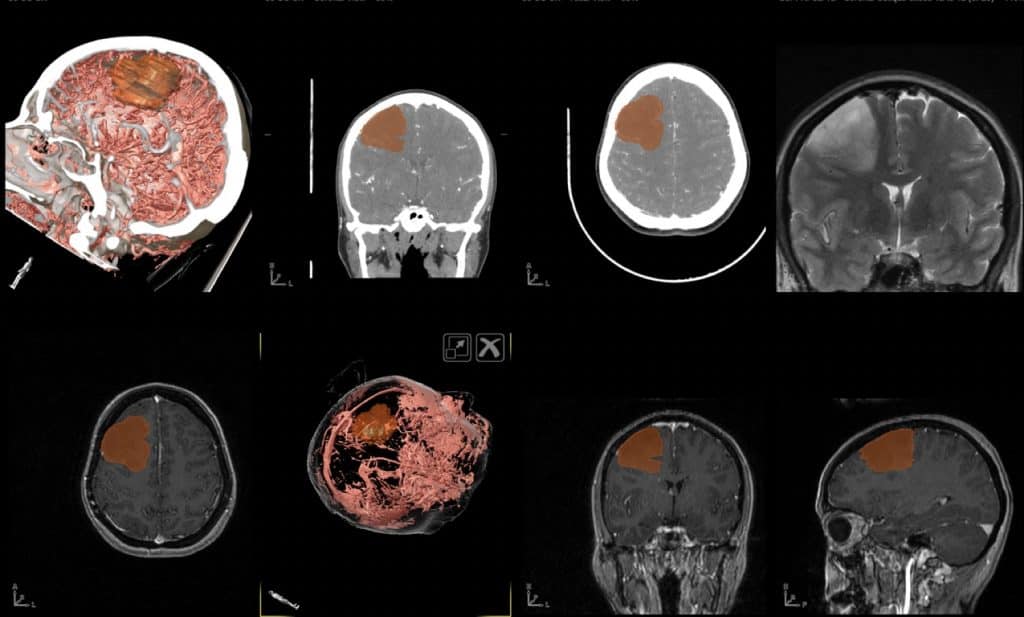
Pero esta regla se altera en la edad pediátrica, en que además hay una serie de entidades típicas de esta edad, que consisten en tumores benignos (astrocitomas pilocíticos) localizados a nivel del nervio óptico-quiasma, cerebelo y tronco cerebral.
3.2.1.3.- CLINICA
El síntoma más frecuente es la epilepsia (40-75 %), sobre todo en los tumores que afectan la corteza cerebral, siendo más rara su presentación en los tumores subcorticales.
El déficit focal se presenta, como primer síntoma, en un 20 % de casos.
La cefalea está presente en un 70 % de los casos y en sólo un 10 % esta cefalea es localizadora. Hasta en un 50 % de los casos aparece edema de papila cuando son explorados.
El resto de los signos exploratorios dependen del déficit ocasionado.
Los astrocitomas talámicos y, sobre todo, los astrocitomas de cerebelo pueden producir hidrocefalia
3.2.1.4.- DIAGNÓSTICO
La posibilidad de un astrocitoma debe ser sospechada en un paciente con crisis epilépticas que aparecen después de los 20 años de edad.
La Rx suele ser negativa, aunque a veces se pueden ver signos de HIC o calcificaciones (8%).
El EEG puede localizar un foco irritativo-lesivo sugerente de lesión tumoral.
La TAC es el método diagnóstico rápido de elección, que después complementará la RM. En los astrocitomas grado I-II aparece zona de hipodensidad (TAC) o hipointensidad (RM), sin edema circundante y sin realce tras la administración de contraste. Los de grado III-IV tienen mayor tendencia a ser hiperdensos (TAC) o isointensos-hiperintensos (RM), con límites cada vez menos homogéneos e infiltrantes a medida que aumenta su agresividad, con zonas irregulares de necrosis en su interior (hipodensas o hipointensas); suelen tener edema circundante y en el 90 % se refuerzan tras la administración de contraste. La presencia de calcificaciones (visibles mejor en la TAC) o de zonas quísticas no es infrecuente.
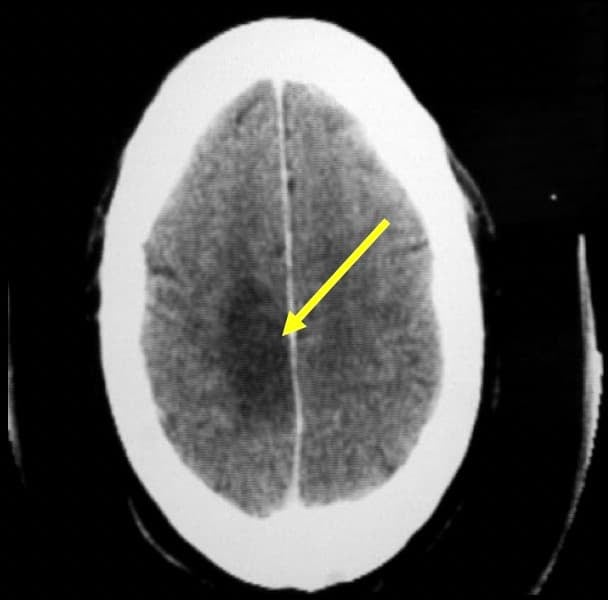
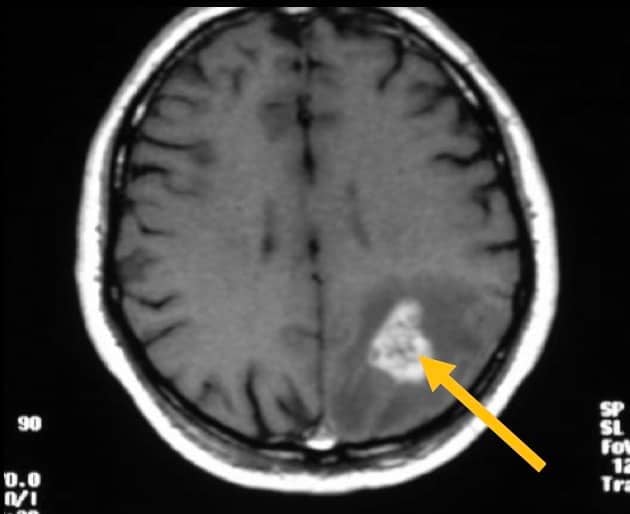
La angiografía tiene poco valor diagnóstico. Los astrocitomas de grado I y II suelen ser avasculares, mientras que a medida que aumenta la agresividad se incrementa la vascularización, con vasos anómalos neoformados, tanto arteriales como venosos.
3.2.1.5.- TRATAMIENTO
En el caso de un posible astrocitoma benigno productor de epilepsia como único síntoma, el tratamiento ha de ser guiado por protocolos de Cirugía de la Epilepsia, con el objetivo de extirpar completamente la lesión y erradicar las crisis.
Según edad y estado general del paciente, la apariencia de agresividad en la RM, la localización profunda o cortical y las posibles áreas funcionales que afecte, se planteará una biopsia estereotáxica como única intervención quirúrgica o, mejor, el intento de extirpación completa. Hoy día, con los recursos a nuestro alcance, es rara la indicación en estos casos de hacer solamente biopsia y se ha de intentar la extirpación más radical posible.
Según el resultado anatomopatológico, el tratamiento se completará con radioterapia y quimioterapia.
Los astrocitomas de grado II tienen una suprevivencia del 30% a los 20 años, los de grado III, la supervivencia a los 10 años es inferior al 30%
3.2.2.- OTROS ASTROCITOMAS
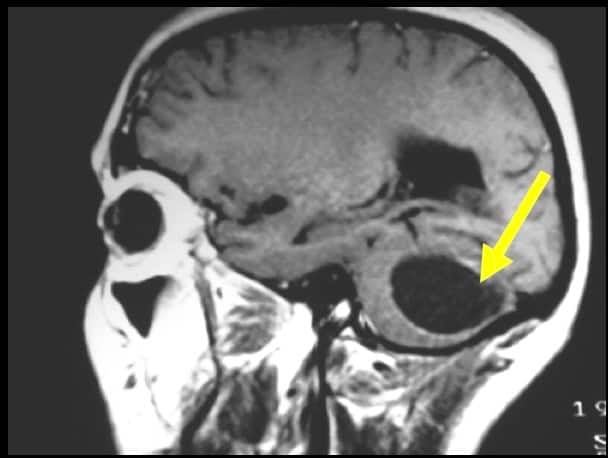
3.2.2.1.- ASTROCITOMA PILOCITICO JUVENIL
Afectan predominantemente a niños y jóvenes y su localización más frecuente es infratentorial. El aspecto en RM es de de lesiones quísticas con un nódulo que capta contraste. En el estudio anatomopatológico son típicas las fibras de Rosenthal. Son tumores de bajo grado y el tratamiento es la extirpación completa con una supervivencia del 100% si la cirugía es radical
3.2.2.2.- ASTROCITOMA SUBEPENDIMARIO DE CELULAS GIGANTES
Tiene mayor incidencia en la infancia y se asocia con la Esclerosis Tuberosa, su localización preferente es periventricular. El tratamiento es quirúrgico
3.2.3.- OLIGODENDROGLIOMA
Estos tumores, derivados de los oligodendrocitos, son raros y se caracterizan por ser generalmente benignos (Grados I-II) y presentar calcificaciones en su interior (el 70%).
Un tercio tienen un componente mixto de células astrocitarias o ependimarias. Hoy en día para su diagnostico histológico y estadiaje es preciso realizar los estudios de codeleccion 1p, 19q para establecer el diagnostico.
3.2.3.1.- INCIDENCIA
Es un tumor de presentación infrecuente (4 % de los gliomas). Es más frecuente en hombres de edad media de 40 años.
3.2.3.2.- LOCALIZACION
En el 90 % de los casos son supratentoriales y más frecuentes en el lóbulo frontal.
3.2.3.3.- CLINICA
Historia larga de 7-8 años de evolución, siendo en un 50 % la epilepsia el síntoma inicial.
Aparece posteriormente cefalea y edema de papila, por el gran tamaño que adquiere el tumor. El déficit neurológico se presenta en un 30 % de los pacientes. Una vez establecido el cuadro el 90 % de los pacientes tienen epilepsia.
3.2.3.4.- DIAGNOSTICO
Rx de cráneo: Es posible visualizar las calcificaciones en un 40-50 % de los casos. Aunque, si en un paciente con epilepsia y de edad media hacemos una Rx y encontramos calcificaciones, lo más posible es que se trate de un astrocitoma más que de un oligodendroglioma, dada la mayor frecuencia del astrocitoma sobre el oligodendroglioma, a pesar de que la frecuencia de presencia de calcificaciones sea menor en el astrocitoma.
En la TAC, más sensible para el calcio, en el 90% de los casos se ven imágenes características de su presencia.
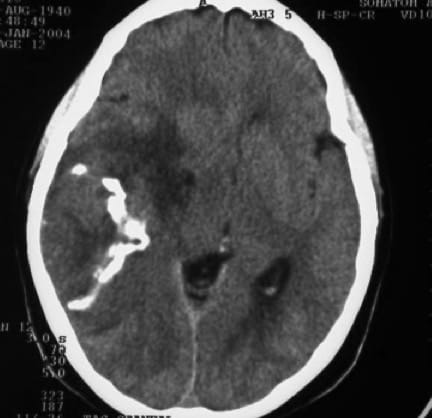
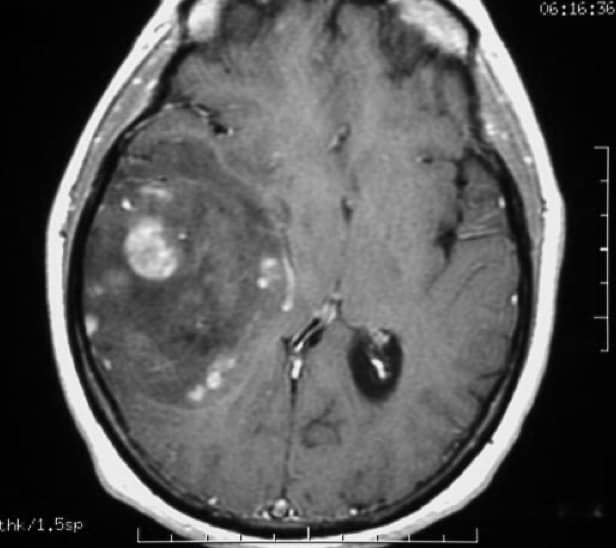
Tanto en la TAC como en la RM la lesión tumoral aparece relativamente circunscrita, de importante tamaño, produciendo desviación de línea media, de apariencia relativamente homogénea, sin edema circundante y que capta contraste ligeramente, todo ello en concordancia con su poca agresividad.
3.2.3.5.- TRATAMIENTO
Es quirúrgico, intentando realizar la extirpación lo más completa posible. Dependiendo de su agresividad histológica, se completa el tratamiento con radioterapia y/o quimioterapia.
3.2.4.- EPENDIMOMA
Lo más característico de estos tumores es que suelen aparecer en edad pediátrica o en jóvenes y cursar solamente con síntomas de hipertensión intracraneal, por la hidrocefalia que producen.
Pueden producir siembras a través del LCR
3.2.4.1.- INCIDENCIA
Constituyen el 5 % de los gliomas. La edad media de presentación es de 20 años, con discreto predominio en varones.
3.2.4.2.- LOCALIZACION
2/3 son infratentoriales, a nivel en el IV ventrículo.
En niños suelen ser infratentoriales y en jóvenes infra o supratentoriales.
3.2.4.3.- CLINICA
La duración de los síntomas es intermedia (hasta 1-1’5 años). Su situación intraventricular hace que sean silentes por mucho tiempo hasta que provocan clínica de HIC por hidrocefalia: papiledema (90%), cefalea (80%), vómitos (75 %), ataxia o vértigo (60%).
Como síntomas de inicio, la epilepsia es rara, así como los déficits neurológicos.
3.2.4.4.- DIAGNOSTICO
TAC y RM: imagen tumoral intraventricular, con límites relativamente homogéneos, con estructura no homogénea en su interior, que captan contraste y pueden llegar a producir edema en las paredes ventriculares donde se asientan. Cursan con hidrocefalia obstructiva univentricular (ependimoma del I o II ventrículos, por obstrucción del agujero de Monro o bloqueo de astas ventriculares), biventricular (ependimoma en el III ventrículo) o triventricular (ependimoma del IV ventrículo).
Al igual que con el meduloblastoma, por la alta posibilidad de siembras, hay que plantear estudio de neuroraquis con resonancia cervico-dorso-lumbar para visualizar dichas siembras y realizar los tratamientos complementarios mas agresivos
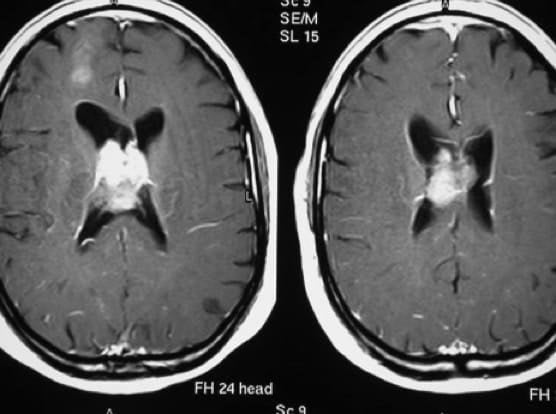
3.2.4.5.- TRATAMIENTO
El tratamiento es quirúrgico con extirpación completa del tumor, si es posible, para evitar las recidivas. En los casos en que la extirpación no sea completa o persista hidrocefalia habrá que actuar sobre esta situación con ventriculostomias endoscópicas o derivaciones de líquido cefalorraquídeo.
Precisan seguimiento y dependiendo de la agresividad completar el tratamiento con radio y/o quimioterapia.
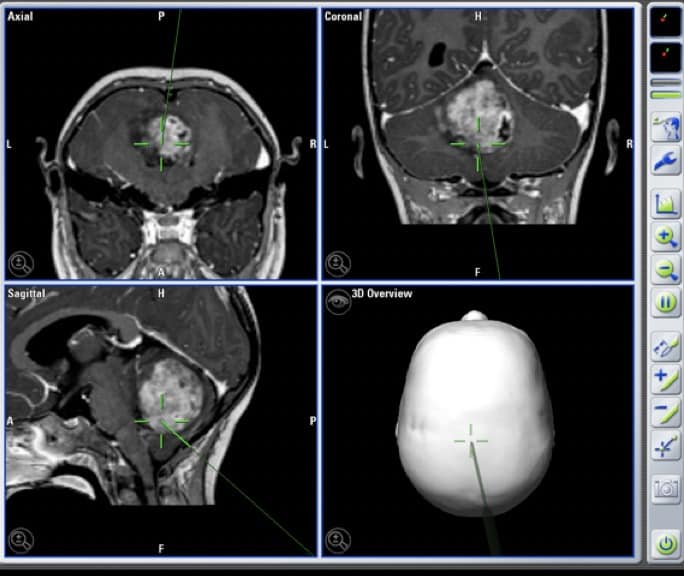
3.2.5.- GLIOBLASTOMA MULTIFORME
Es el tumor cerebral más frecuente y uno de los más agresivos del organismo. Hasta el momento sólo se consigue prolongar la vida del paciente con los tratamientos actuales, aunque cada vez con mejores niveles de calidad de vida.
3.2.5.1.- INCIDENCIA
Es el tumor cerebral primitivo más frecuente, representando el 25 % de los tumores cerebrales y el 50 % de todos los gliomas.
La edad de aparición tiene un pico entre los 40-60 años. Sin embargo, la incidencia parece que va aumentando con la edad. Son muy raros en niños. Son más frecuentes en varones. Pueden ser de novo (más agresivos) o por progresión de un astrocitoma anaplásico.
3.2.5.2.- CARACTERÍSTICAS
Suele ser altamente infiltrante en los 4/5 de los casos, o aparentemente circunscrito en el 1/5 restante. Puede ser quístico (40 %), sólido (60%), hemorrágico (40%) y con zonas necróticas en su interior (50%). No es infrecuente que curse con un cuadro de hemorragia espontánea (20%).
Las zonas de mayor frecuencia de presentación son los lóbulos frontal y temporal, así como en el cuerpo calloso (por donde se difunde), adoptando una imagen característica en alas de mariposa.
El 20 % de ellos aparecen con focos múltiples, pero realmente solo el 2 % se pude considerar como un tumor multicéntrico.
A pesar de su agresividad, muy raras veces produce siembras vía LCR que sean sintomáticas (cuando alcanzan el epéndima y el ventrículo), aunque se pueden encontrar siembras asintomáticas al realizar la autopsia. Quizás debido a la corta expectativa de vida no les da tiempo a desarrollarse. Se pueden detectar células en el LCR en un 40% de los casos.
Las metástasis fuera del SNC de los tumores primitivos son excepcionales (en el 0,4 % de los casos) pero, en caso de producirse, en el 66 % de los casos son por glioblastoma multiforme.
3.2.5.3.- CLINICA
La presentación de los síntomas se hace en muy breve plazo, dado el alto índice de crecimiento tumoral. Así, el 30 % de los pacientes se presentan con una duración de los síntomas menor de un mes, el 60 % menor de tres meses y sólo en el 7 % de los pacientes se pueden ver síntomas con duración mayor de 2 años.
La cefalea se presenta en el 70 % de los pacientes y es el síntoma inicial en el 40% de los casos.
La disminución de fuerza se presenta como síntoma de inicio en el 3 % de los pacientes y se encuentra en el 43 % de ellos cuando se hace el diagnóstico.
Los trastornos mentales y de comportamiento también son un signo frecuente de presentación (45 %).
Sin embargo, las crisis epilépticas son raras como síntoma inicial: 15 %.
La evolución de todos los síntomas es muy rápida, añadiéndose progresivamente clínica de HIC y síntomas de déficits focales.
3.2.5.4.- DIAGNOSTICO
En la TAC y RM, se aprecia una masa tumoral de importante tamaño, con límites imprecisos, que capta contraste de forma no homogénea y tiene abundantes zonas de necrosis en el interior, junto con edema importante digitiforme peritumoral. La gran masa produce importantes distorsiones de las estructuras encefálicas.
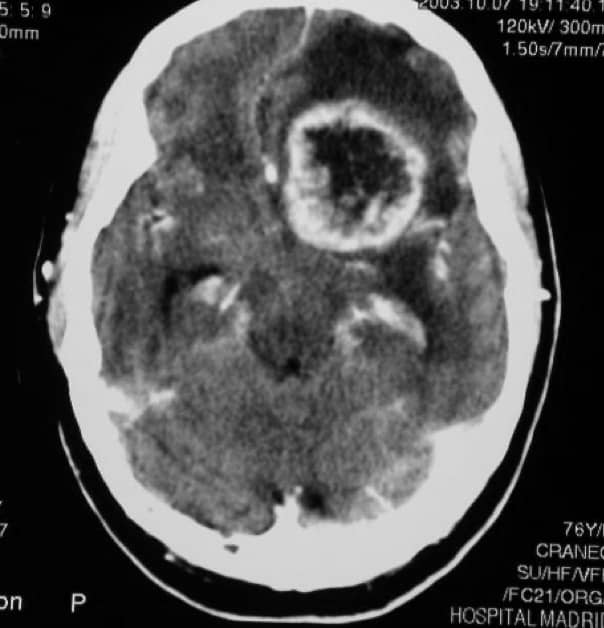
3.2.5.5.- TRATAMIENTO
Ante una imagen en la TAC y RM sugerente de glioma maligno no es coherente dejar al paciente sin actuación terapéutica alguna. Excepto en muy raras ocasiones en que la edad muy avanzada o el mal estado general así lo aconsejen. Una de las razones es que pudiera tratarse de un absceso cerebral (que pueden dar imágenes cerebrales muy similares); aunque esto puede suceder en casos excepcionales, todos tenemos recuerdos de algunos pacientes a los que se les ha salvado la vida gracias a una biopsia o una craneotomía, encontrando inesperadamente un proceso benigno en lugar de la tumoración maligna.
Con la inhibición quirúrgica estamos impidiendo que al paciente se le puedan ofrecer las alternativas terapéuticas actuales, que pueden prolongar la vida útil, aparte de conocer su propia expectativa de vida. En este sentido, no es infrecuente que por la imagen pensemos en un grado de malignidad muy alto y, sin embargo, tras el estudio anatomopatológico, nos encontramos con una menor agresividad biológica tumoral.
Aunque no puede ser una razón a poner en la balanza de una difícil decisión, hemos de saber que, si no biopsiamos las lesiones, nuestro desconocimiento va a seguir siendo importante sobre muchos aspectos: diagnóstico e incidencia real de los diferentes tipos de tumores; pronóstico de cada uno de ellos y respuesta a las nuevas pautas terapeúticas que continuamente van surgiendo.
La disyuntiva sobre si hacer biopsia o intentar la resección macroscópica lo más amplia posible dependerá de la localización tumoral y áreas funcionales circundantes. Hay que tener en cuenta que se consigue mayor expectativa de vida en tumores a los que se les realizado una extirpación radical que a los que sólo se les ha realizado biopsia.
Para facilitar una resección del más del 90% de la masa tumoral, sin déficits neurológicos, tenemos actualmente tres herramientas a nuestra disposición:
1. El neuronavegador, que nos permite visualizar la tumoración, sus limites y su relación con los principales tractos y circunvoluciones a respetar.
2. La posibilidad de administrar una medicación [acido 5 aminolevulinico (5-ALA, comercializado como Gliolan)] unas horas antes de la intervención quirúrgica que va a ser absorbida por células tumorales agresivas. Esta sustancia tiene unas características de fluorescencia que permite ver, en microscopios adecuados, el tejido tumoral de color rojo, mientras que el tejido cerebral normal está en azul. En esta página web se pueden ver varios casos de intervenciones en que se puede apreciar esta fluorescencia. Esto permite la resección de tejido que a la luz normal del microscopio parece normal, pero que está infiltrado por el tumor.
3. La colaboración de Neurofisiología y Neuropsicología durante la intervención quirúrgica, que permite preservar las zonas funcionales neurológicamente importantes.
Posteriormente se ha de planear una actuación conjunta de radioterapia (convencional o estereotáxica fraccionada) y quimioterapia.
3.2.5.6.- PRONOSTICO
Si no se realiza actuación terapéutica alguna o se hace solamente una biopsia estereotáxica, la media de expectativa de vida no supera los 6 meses desde que se realiza el diagnóstico.
Por el contrario, la resección radical de más del 90% de masa tumoral visible en RM, más tratamiento coadyuvante de radioterapia y quimioterapia, está consiguiendo supervivencias superiores a los 2 años en un porcentaje creciente, manteniendo una buena calidad de vida. Todo esto va a ser posible y va a depender, por tanto, de la capacidad quirúrgica, localización tumoral y edad del paciente,
3.2.6.- GENERALIDADES DEL TRATAMIENTO DE LOS GLIOMAS
El tratamiento ideal de cualquier tumor es la extirpación radical. Esto no siempre es posible por estar próximos o implicar a áreas elocuentes.
Hoy en día las técnicas de RM como la espectroscopía pueden dar una aproximación al grado de agresividad del tumor así como a su posible diagnóstico. Pero los avances en los tratamientos complementarios precisan de un estudio histológico y de marcadores, para poder definir y clasificar mejor el tumor, así como los tratamientos complementarios.
Como tratamientos previos a la intervención quirúrgica y en el postoperatorio inmediato, para control del edema cerebral producido por el tumor, se utilizan los corticoides y en concreto la dexametasona. También se deben administrar anticomiciales para prevenir las crisis comiciales.
Es excepcional que no se pueda intentar una resección radical del tumor, debido a su proximidad a áreas elocuentes. Por lo que la frecuencia de las biopsias estereotácticas está disminuyendo.
Por consiguiente, el planteamiento actual es intentar una resección radical de cualquier tipo de glioma, con bajo riesgo de lesión neurológica. Como hemos referido anteriormente, esto se puede conseguir gracias a las técnicas de neuronavegación, fluorescencia y control neurofisiológico-neuropsicológico intraoperatorios.
Una controversia actual es si llevar a cabo este tipo de intervenciones con el paciente despierto. Esto llama la atención a las personas fuera del mundo neuroquirúrgico. Pero ya era la técnica de elección en los años 40, dado el riesgo entonces de la anestesia general.
Así Penfield y Jasper pudieron legar una metodología para estudio del paciente epiléptico, con descripción de muchas zonas neurológicamente importantes (recuérdese el homúnculo de Penfield, por ejemplo).
La necesidad absoluta de mantener al paciente despierto es cuando la zona a intervenir tiene relación con zonas del lenguaje.
La ayuda de la neuropsicología está permitiendo, además, estudiar otras zonas aparentemente no tan importantes, en cuanto a déficits y calidad de vida, pero que nos están ayudando a comprender mejor la estructura-función cerebral. Pero esto debería quedar limitado a los neurocirujanos y equipos que dispongan de esta capacidad de estudio.
Por otro lado, la monitorización neurofisiológica intraoperatoria (MNIO) permite controlar funciones, aunque el paciente esté dormido: Motora (PEMs), sensitiva (PESS), visual (PEVs) o auditiva (PEATs). Lo que permite que el paciente esté en una situación de menor estrés y que la valoración intraoperatoria sea más objetiva, no dependa de la respuesta del paciente.
En definitiva, ha de ser el neurocirujano y sus recursos, junto al paciente, quienes elijan cómo llevar a cabo la intervención quirúrgica.
Tras la cirugía puede ser necesario completar el tratamiento con técnicas de radioterapia y quimioterapia en aquellos tumores más agresivos.
En gliomas de alto grado se utiliza el esquema de Stupp que consiste en el tratamiento de radioterapia sobre el lecho del tumor y con un margen de seguridad a dosis de 60 Gy repartidos en 25-30 sesiones administrando de forma concomitante Temozolamida.
Una vez finalizado el tratamiento de radioterapia se realizan ciclos de Temozolamida durante 5 días cada 4 semanas hasta completar al menos 6 ciclos. La Temozolamida es de administración oral y con buena tolerancia. Precisa control analítico por la posibilidad de provocar plaquetopenia.
Otros quimioterápicos que se están utilizando para el tratamientos de los gliomas, en el caso de recidivas, es el Bevacizumab (Avastin) o la Fotemustina. En algunos casos se está volviendo a las pautas clásicas de las asociaciones de Lomustina (CCNU), Vincristina y Procarbacina. También se han utilizado durante años la implantación de quimioterapia en el lecho quirúrgico con polimeros de Carmustina, tratamiento actualmente poco habitual al no haber aportado grandes beneficios.
Ante un tumor glial de bajo grado y con resección completa, en el tratamiento postquirúrgico se tiende a la observación y vigilancia. Si la resección no es completa se puede asociar el tratamiento de radio y/o quimioterapia
En tumores de grado III y alto grado como los Glioblastomas hay que ser más agresivos y completar el tratamiento quirúrgico con la radioterapia y quimioterapia. En casos de recidivas, si es posible, se plantea una reintervención para prolongar la supervivencia.


muy buena la informacion, agradesco alos doctores por un buen trabajo
Excelente
Muy buena información clínica y académica
Que continúen los éxitos
Gracias por su tiempo