
MALFORMACIONES DEL SISTEMA NERVIOSO
Tema II: Temario de Neurocirugía 2020 para alumnos de Facultad de Medicina de la UAM: 2020
Principales Malformaciones del Sistema Nervioso
Facultad de Medicina de la Universidad Autónoma de Madrid.
Directores:
MALFORMACIONES DEL SISTEMA NERVIOSO – CONCEPTO
Son un defecto anatómico evidente desde el nacimiento, causado por un insulto al embrión durante el embarazo.
ETIOLOGÍA
Las causas son muy variadas, pudiendo ser clasificadas en:
a.- Hereditarias
b.- Ambientales:
– factores biológicos generales: edad de los padres, incompatibilidad sanguínea…
– deficiencias nutricionales
– infecciones maternas. rubeola, toxoplasmosis
– acciones hormonales: andrógenos,..
– efectos físicos: Rx
– efectos químicos: drogas, fármacos…
INCIDENCIA
El riesgo de malformaciones congénitas es, en general, de aproximadamente un 2% de los nacimientos, mientras que el porcentaje de malformaciones del Sistema Nervioso Central (SNC) es del 2,66 por mil. Es difícil conocer la existencia de malformaciones en abortos espontáneos y su porcentaje puede ser superior al 30 %. La administración de acido fólico en las embarazadas ha reducido el porcentaje también de malformaciones. Son responsables del 75% de las muertes fetales y el 40% de las muertes en el primer año de vida. También suponen una importante causa de minusvalía infantil.
Embriología
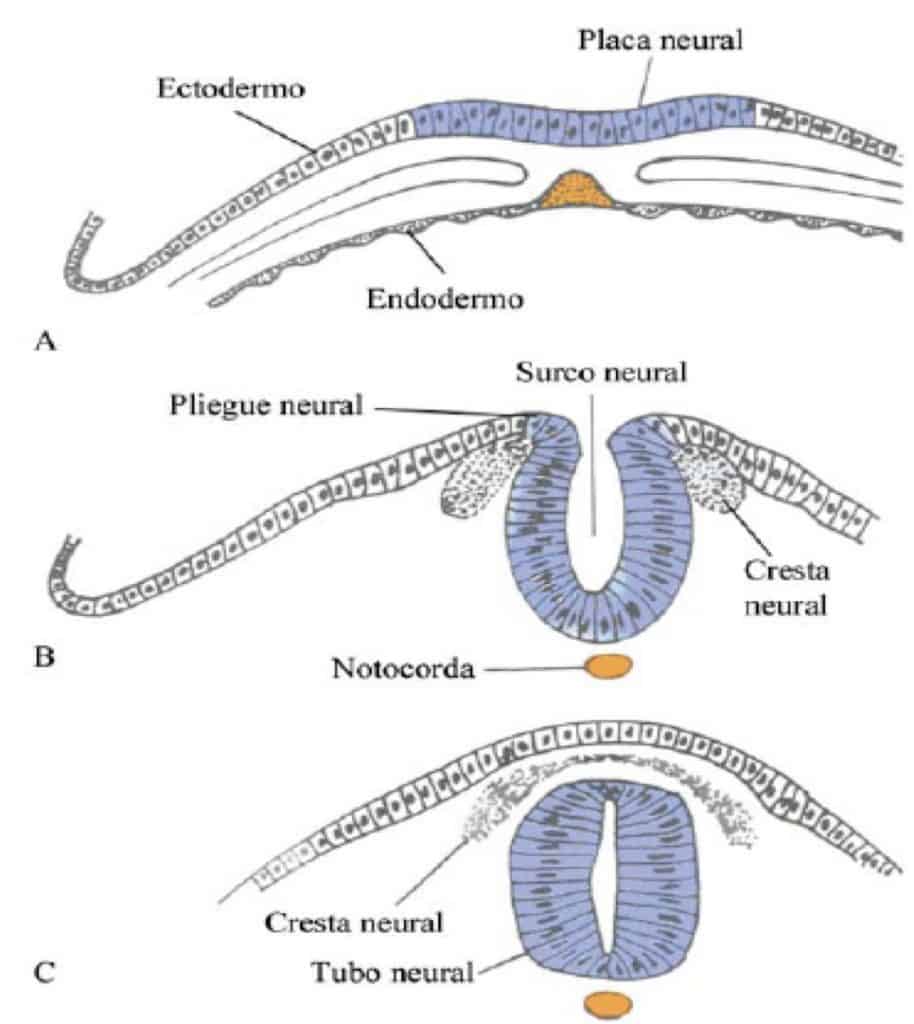
Recordemos que el Sistema Nervioso se forma a partir de la placa neural: Pliegues neurales, tubo neural y cresta neural. El tubo neural va a formar el encéfalo y la médula espinal y de la cresta neural se dará origen al sistema nervioso periférico y autónomo.
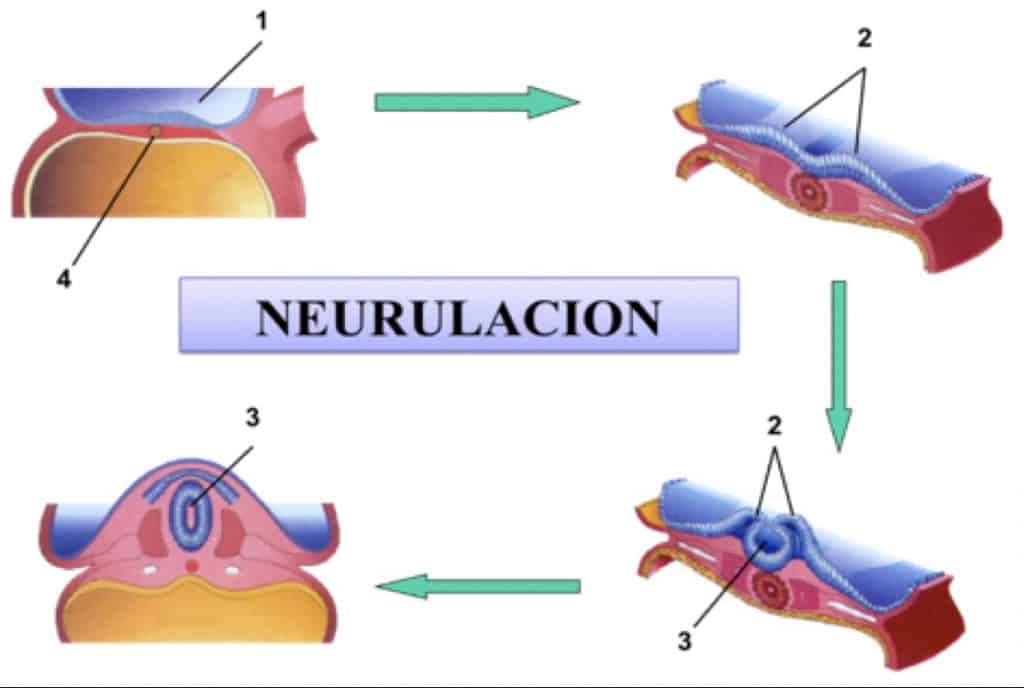
La neurulación comienza el día 22-23. 2/3 formarán el encéfalo y 1/3 la médula espinal
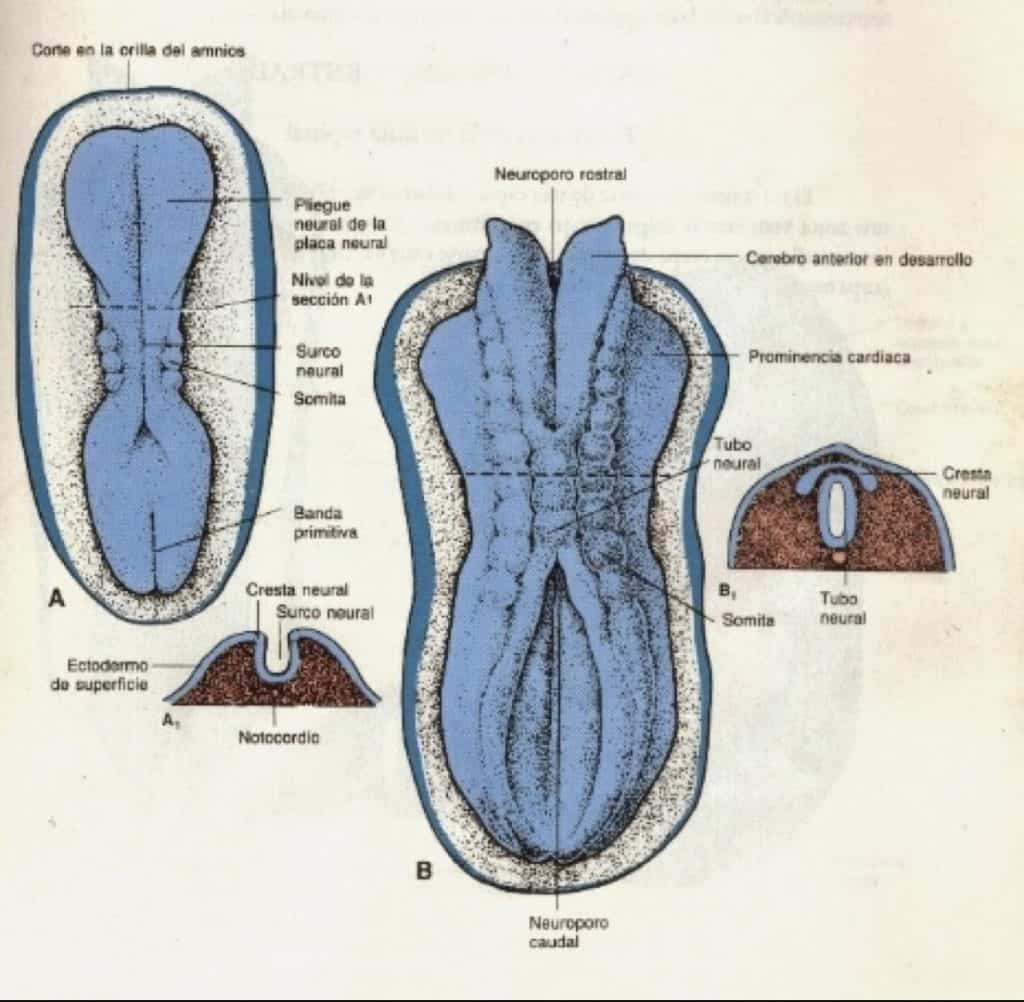
La fusión de los pliegues neurales se produce en dirección craneal y caudal quedando unos orificios en los extremos:
– el neuroporo anterior: cierra el día 25
– el neuroporo posterior: cierra el día 27
Coincide con la aparición de la circulación vascular para el tubo neural. Las paredes del tubo neural se engrosan y forman el encéfalo y la médula. El conducto neural forma el sistema ventricular y el conducto central medular.
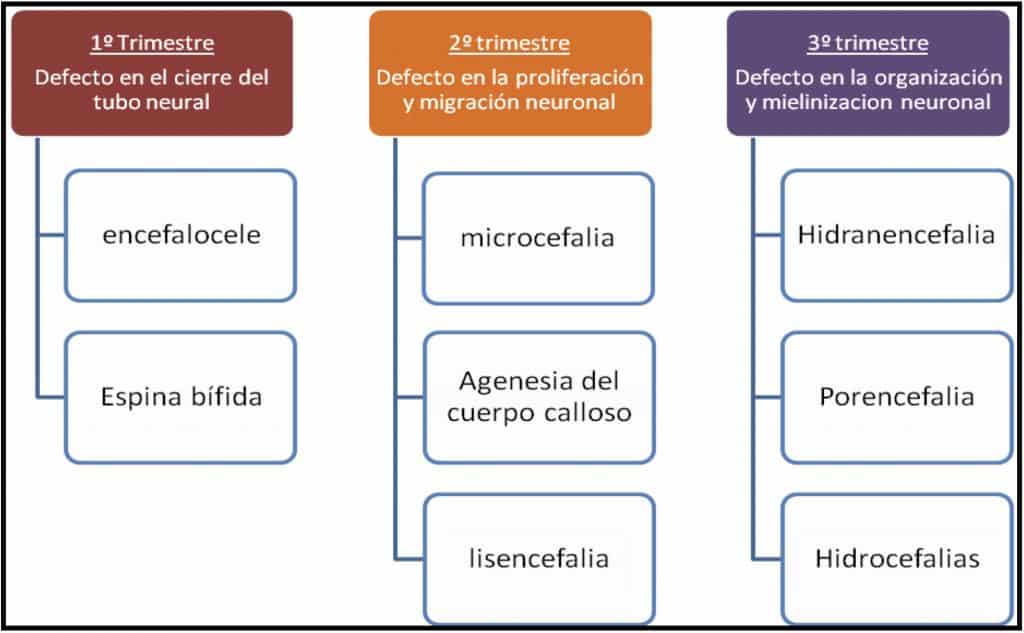
Si dividimos el desarrollo embrionario en tres períodos, muy esquemáticamente, las causas incidirían en las siguientes etapas de formación del SNC:
– 1er trimestre: se dificulta la formación del tubo neural: disrafismo craneal o espinal
– 2º trimestre: alteraciones en la proliferación y migración neuronal.
– 3er trimestre: alteración en la organización neuronal y en el proceso de mielinización.
A medida que la noxa actúa más precozmente, la malformación va a ser más grave e incompatible con la vida
PARTE I.- MALFORMACIONES CONGENITAS CRANEO-ENCEFALICAS
CLASIFICACION – MALFORMACIONES DEL SISTEMA NERVIOSO
Las alteraciones congénitas se pueden presentar a nivel de piel, cráneo o en el propio encéfalo. Las más importantes, desde el punto de vista neuroquirúrgico (por su posibilidad de reparación mediante una intervención quirúrgica), pertenecen al grupo de alteraciones en el cierre y constitución del tubo neural y las capas que lo envuelve y protegen:

A.- ALTERACIONES EN LA FORMACION DEL TUBO NEURAL
I.- SINUS DERMICO CRANEAL
Defecto de las cubiertas por anomalía en su inducción dorsal durante la 3ª-4ª semana del desarrollo embrionario, consistente en que en algún punto no se despega por completo el tubo neural del ectodermo y queda una invaginación cutánea que forma un tracto más o menos permeable, revestido por epitelio escamoso estratificado, que conecta epidermis con tejido nervioso.
Se localiza en línea media o en su proximidad y pueden aparecer desde nasion hasta el coxis, aunque la localización más frecuente es a nivel de los neuroporos anterior y posterior: región occipital y región lumbosacra.
A nivel craneal, el más frecuente es el sinus dérmico occipital. Puede ser de tamaño muy variable: pequeño y terminar en el tejido subcutáneo, o grandes y llegar el trayecto hasta el IV ventrículo. Pueden presentar además quistes o tumores de inclusión (dermoides o epidermoides)
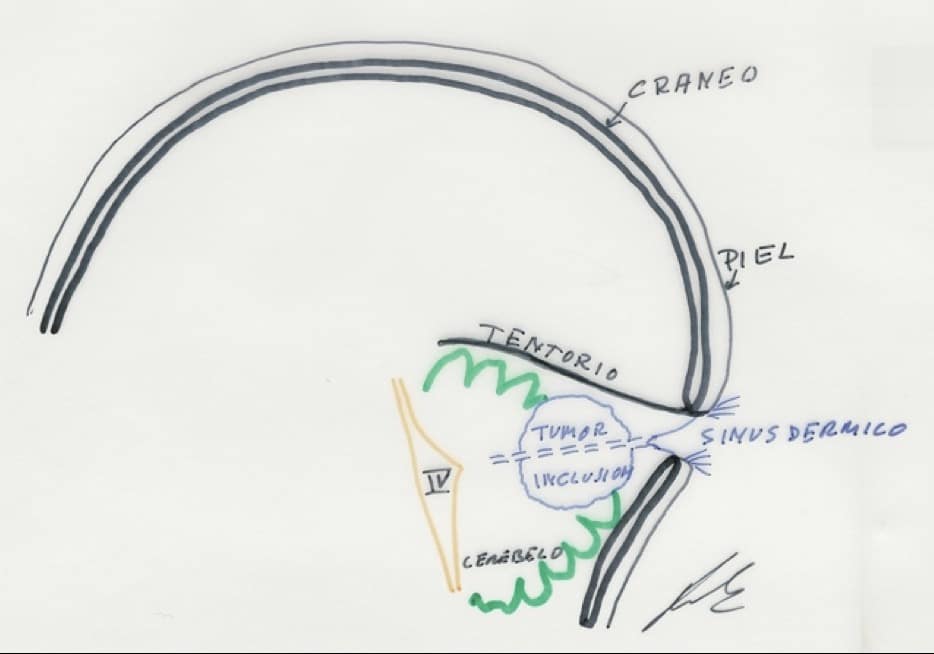
CLINICA.- Se observan alteraciones en la piel, como pequeñas depresiones, acompañadas de pelos, manchas color rojo vinoso… Permanecen asintomáticos hasta que se infectan y producen meningitis por staphilococus aureus y abscesos de repetición. Rara vez pueden llegar a producir cuadros de meningitis aséptica o química por rotura del quiste epidermoide en espacio subaracnoideo. Si los tumores de inclusión alcanzan gran tamaño, pueden dar clínica por efecto de masa y comportarse como proceso expansivo.
DIAGNOSTICO.- Hay que sospecharlo y explorar al niño en la región occipital, sobre todo si es un niño con un cuadro de meningitis de causa desconocida. La Resonancia Magnética es el estudio de elección y completa el proceso diagnóstico. Visualiza el tumor de inclusión e incluso el tracto.
TRATAMIENTO.- Extirpación completa del tracto incluyendo el tumor de inclusión.
II.- APLASIA DE CUTIS:
Defecto cutáneo de piel sobre el vértex a nivel de línea media y próximo a la fontanela posterior. Generalmente solo afecta a piel, pero también puede incluir la gálea, el hueso y la duramadre
El tratamiento es quirúrgico con reparación de las capas afectadas
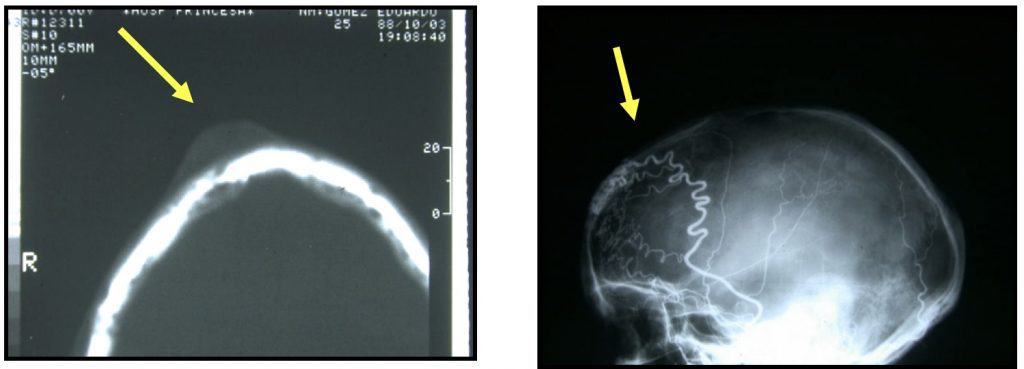
III.- ANEURISMA CIRSOIDEO
Es en realidad una malformación arteriovenosa cutánea, que se visualiza a través de la piel.
CLINICA.- El paciente, aparte de la molestia estética de piel enrojecida con huellas vasculares sinusoidales, refiere sensación de ruido o de latido.
DIAGNOSTICO.- Se hace fácilmente por inspección. El TAC, RM y/o angiografía completan el estudio (Fig. 6).
TRATAMIENTO.- En general, se ha de combinar el esfuerzo del neurorradiólogo intervencionista, para la embolización y reducción del tamaño de la malformación, con la extirpación quirúrgica posterior.
IV.- SINUS PERICRANII
Es una conexión de un seno venoso intracraneal (por lo general del seno longitudinal superior) con una bolsa venosa en cuero cabelludo, por encima del hueso (Fig. 7).
CLINICA.- Da la cara como una tumoración blanda, no dolorosa, que aumenta en posturas en que se incrementa la presión venosa intracraneal (maniobras de Valsalva, acostarse y, sobre todo, bajar la cabeza).
DIAGNOSTICO.- La angiografía puede ser negativa. Por lo que hay que sospecharlo e inyectar el contraste tras puncionar esta bolsa. Se ve cómo se rellena la bolsa y su drenaje al seno venoso intracraneal.
TRATAMIENTO.- Hay que realizar una craneotomía y abordar las conexiones intracraneales, para cerrar la comunicación entre seno y bolsa venosa subcutánea.
V.- ANENCEFALIA
Es la ausencia de bóveda craneal y cubiertas, se observa una masa de neuroglia, hemorrágica expuesta. Es letal falleciendo en días o semanas
VI.- DEFECTO OSEO CONGÉNITO
La localización más frecuente es a nivel parietal
El tratamiento es quirúrgico si es muy grande
VII.- CRANEOESTENOSIS O CRANEOSINOSTOSIS.
Consiste en el cierre prematuro de una o varias suturas craneales. Su incidencia es de 1/2000 niños, predominando en un 80 % en varones.
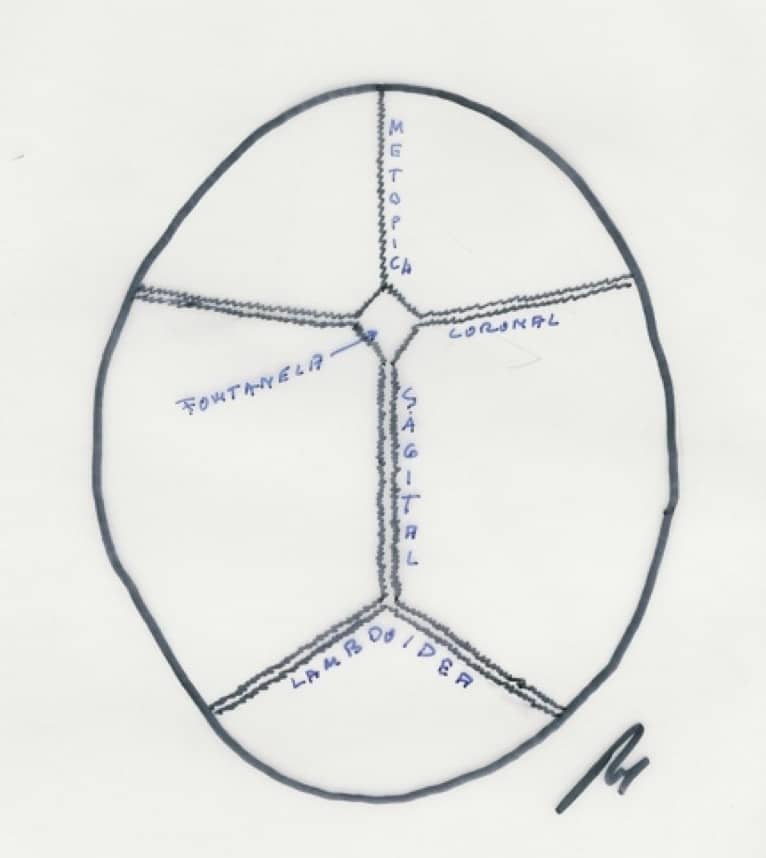
El cierre normal de las suturas se produce en las siguientes etapas: para la sutura metópica, antes del nacimiento; la fontanela posterior se cierra a los 3 meses y la anterior alrededor de los 18 meses. Todas las suturas comienzan a calcificarse a partir de los 8 años y después de los 12 años no se separan aunque exista un cuadro de hipertensión intracraneal
ETIOLOGÍA
Las craneoestenosis pueden ser primarias, presentes antes del nacimiento; o secundarias, debido más frecuentemente a una falta de desarrollo cerebral (microcefalia), raquitismo o tras colocación de derivaciones de LCR en hidrocefalias.
PATOGENIA
Si el cierre precoz se produce solamente a nivel de una sola sutura, se va a producir una deformidad estética. Pero si ya son varias las suturas afectadas, se impide el aumento progresivo del cerebro, generando al principio un cuadro de hipertensión intracraneal para, posteriormente, dificultar el desarrollo cerebral normal, con el consiguiente déficit intelectual o incluso oligofrenia.
CLASIFICACION
De acuerdo con las suturas que sufren el cierre precoz, se clasifican las craneoestenosis de acuerdo a la deformidad craneal que producen:
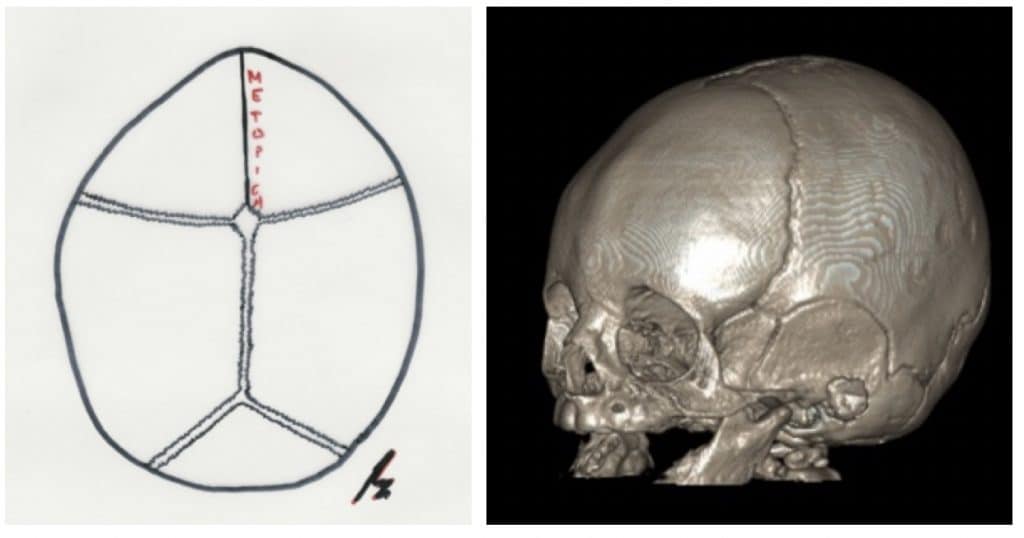
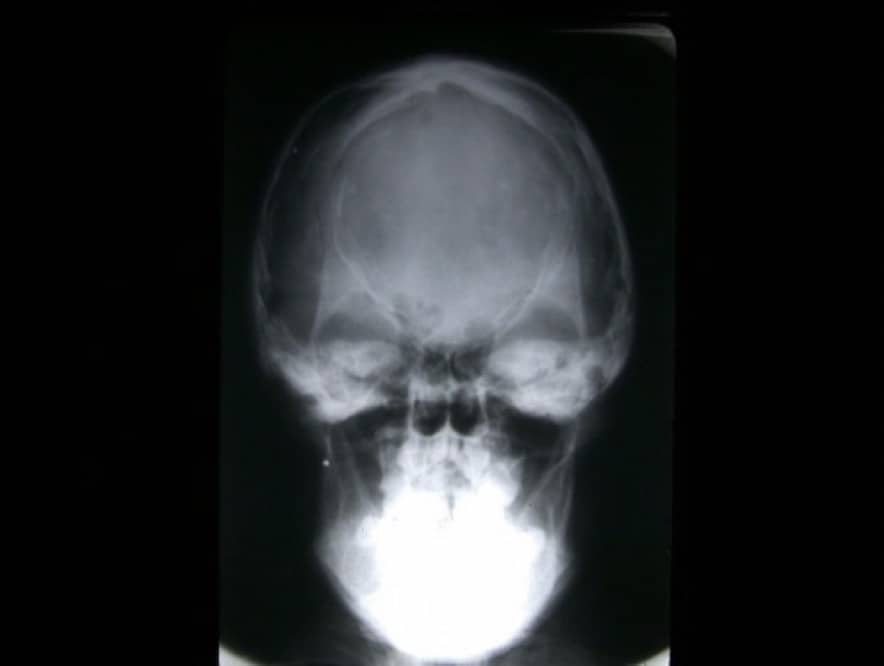
Trigonocefalia: La produce el cierre de la sutura metópica. Suponen el 5-16 % de todas las craneoestenosis. Los niños tienen la frente estrecha y afilada. El nombre lo toman del aspecto de proa de un barco (Fig. 10).
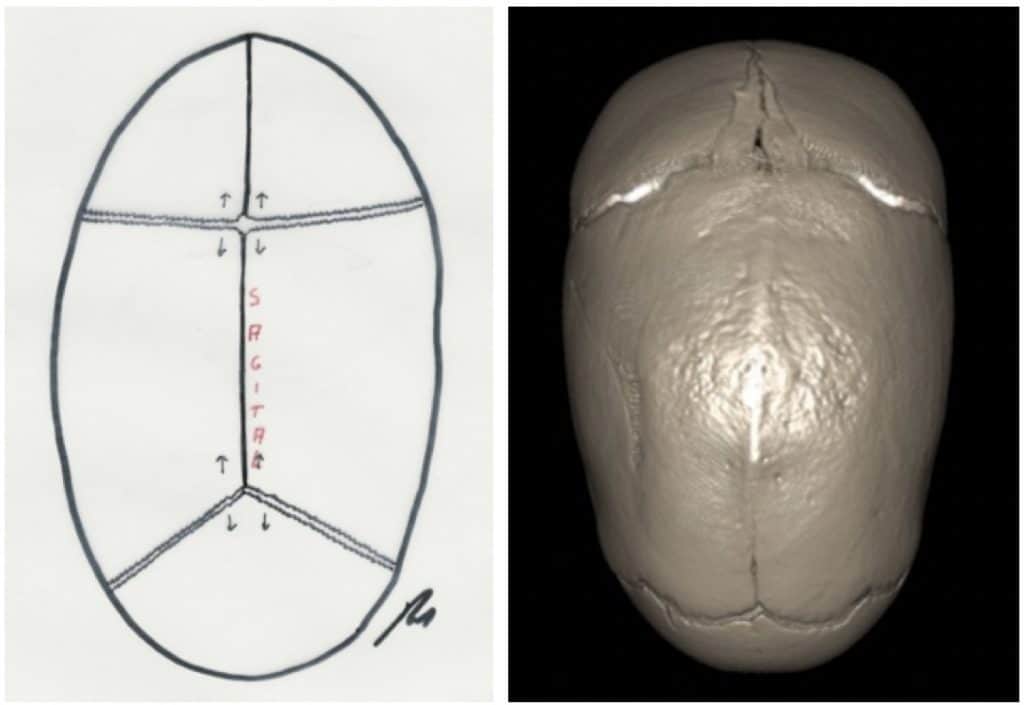
Escafocefalia: Es el cierre de la sutura sagital. Es la deformidad más frecuente (50%). El cráneo adopta la forma de la quilla de un barco (de ahí su nombre), con un aumento del diámetro fronto-occipital (también se denomina dolicocefalia) y una disminución del diámetro biparietal (Fig.11)
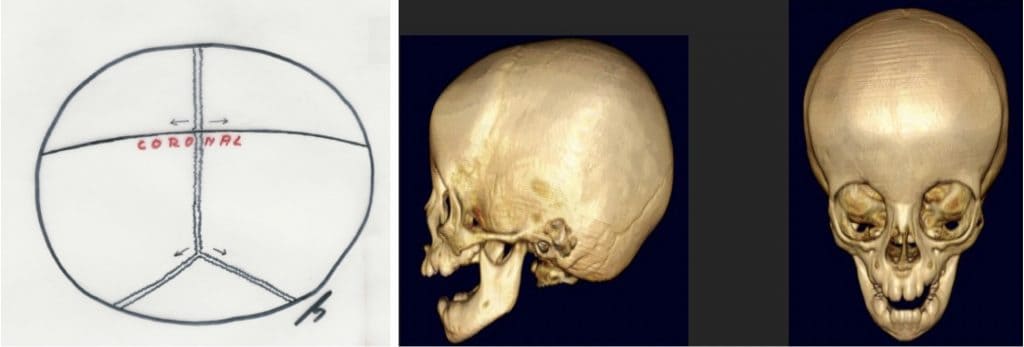
Braquicefalia, acrocefalia: Se debe al cierre de la sutura coronal y su frecuencia de presentación es del 7-15 %. Es lo contrario a la anterior, con disminución del diámetro fronto-occipital y aumento del bifrontal. La cabeza puede adoptar forma de torre al crecer el cráneo hacia arriba (turricefalia) (Fig.12).
Oxicefalia: cierre de varias suturas, coronal y sagital, es un cráneo picudo. Presenta problemas graves de desarrollo cerebral, si no es reparado de forma rápida.
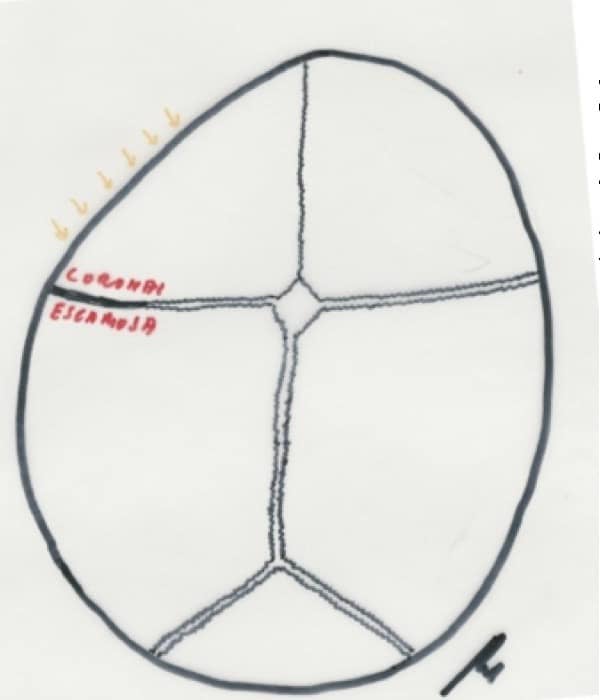
Plagiocefalia: Es un cráneo asimétrico por cierre de un lado de la sutura coronal y la sutura escamosa temporal adyacente. Su frecuencia es del 8-18%. El aspecto de la cabeza es de estar como aplastada en una de las regiones fronto-temporales. (Fig. 13). En las dos ultimas décadas se están produciendo deformidades del cráneo sobre todo a nivel parietal o occipital con cuadros sugestivos de plagiocefalia que, sin embargo, se corresponden con deformidades posicionales del lactante por la tendencia a mantener a los lactantes en decúbito supino para evitar la muerte súbita. Esta deformación no es congénita y el tratamiento es posicional o incluso rehabilitador
Hay otras alteraciones más graves, en las que se asocian el cierre de múltiples suturas con anomalías craneofaciales. Entre éstas destaca la disostosis craneofacial o enfermedad de Crouzon y el Síndrome de Apert o acrocefalosindactilia (Fig. 14).
DIAGNOSTICO DE CRANEOESTENOSIS
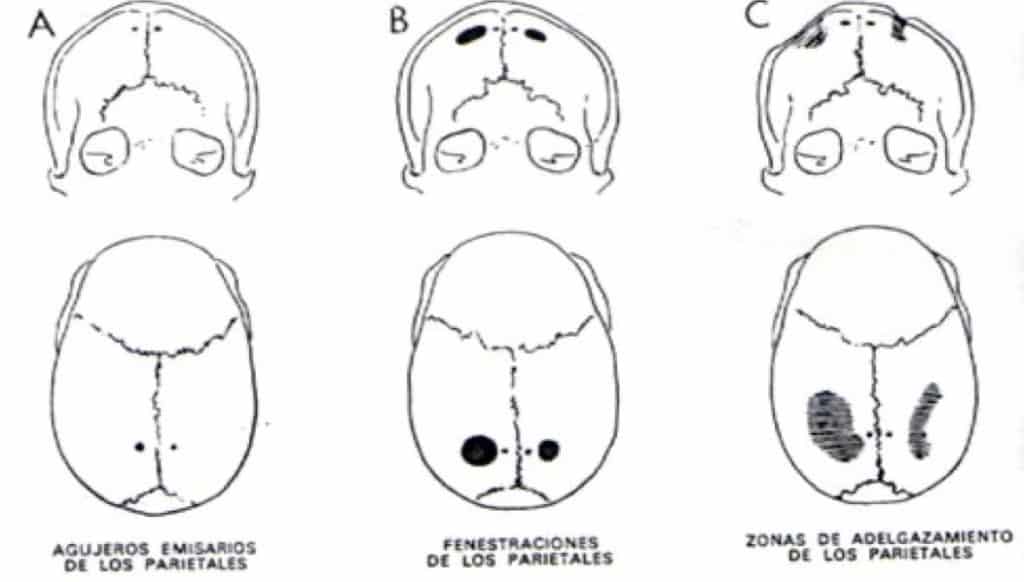
Se realiza tras la inspección del recién nacido. La palpación de la sutura permite apreciar un relieve óseo, duro, lineal a todo lo largo de la sutura cerrada. Actualmente el TAC 3-D permite tener un diagnóstico de certeza.
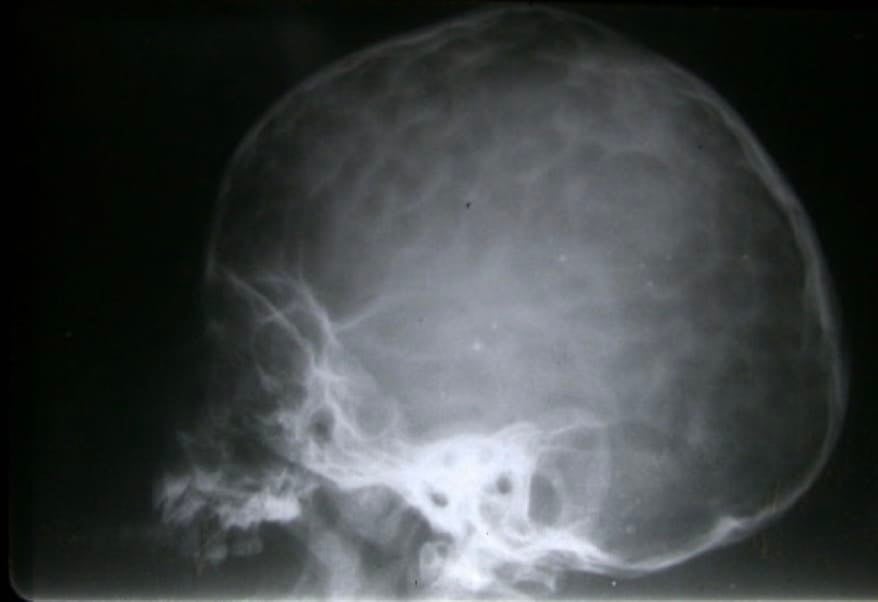
Ya cada vez es más raro que diagnostiquemos una craneoestenosis en edades tardías. En estos casos podemos ver incluso huella digitiformes en las Rx de cráneo, señal de haber existido una hipertensión intracraneal (HIC) (Fig.15). Cuando el niño es mayor de 4-6 años, el problema se plantea sobre si la apertura de las suturas va a ser útil, en cuanto a permitirle un mejor desarrollo intelectual. En caso de duda, es muy útil monitorizar la presión intracraneal a lo largo de 24-48 horas. En caso de observar signos de HIC, se procedería a abrir las suturas, para dejar al cerebro crecer y evolucionar.
DIAGNOSTICO DIFERENCIAL
Hay que hacer el diagnóstico diferencial con la microcefalia secundaria a un menor crecimiento del cerebro. En este caso las suturas están cerradas, pero no precisan su apertura. Suelen ser niños con mayor deterioro intelectual y no presentar asimetrías en la forma del cráneo, aparte de que la RM puede mostrar imágenes de encéfalos dismórficos.
TRATAMIENTO>
El tratamiento quirúrgico consiste en la apertura de las suturas. En los últimos años los avances en las técnicas endoscópicas pueden permitir resecciones de suturas mediante esta técnica, si se realiza en los primeros meses.
Si son varias las suturas afectadas a veces es preciso realizar craniectomias y remodelación de los fragmentos óseos para obtener el efecto estético deseado. Si se sobreañaden anomalías faciales, se requiere combinar las técnicas neuroquirúrgicas con equipos de cirugía máxilo-facial.
El tratamiento quirúrgico se debe realizar preferiblemente antes de los seis primeros meses de vida, puesto que el volumen cerebral en estos meses se va a incrementar en un 80 %. Pero, excepto que el cierre de las suturas sea global, es aconsejable esperar al menos 1 mes de vida para permitirle al niño el suficiente nivel de desarrollo como para soportar una cirugía en la que ineludiblemente pierden sangre
VIII.- ENCEFALOCELE
Es un defecto en el cierre del neuroporo anterior, que se produce durante la cuarta semana de embarazo. El contenido cerebral hace protrusión y se hernia a través del defecto craneal. Su incidencia de 1/2000 nacimientos y pueden representar hasta un 10% de las malformaciones del Sistema Nervioso Central.
CLASIFICACION
De acuerdo con el contenido, los encefaloceles pueden ser:
1.- Meningocele: Sólo herniación de meninges, con contenido de LCR en la tumoración
2.- Encefalomeningocele: Se hernia tejido cerebral, aparte de meninges.
3.- Hidroencefalomeningocele: La malformación es más grave, con salida de una cantidad importante de tejido cerebral (que incluye ventrículo) y meninges. (Fig.16).
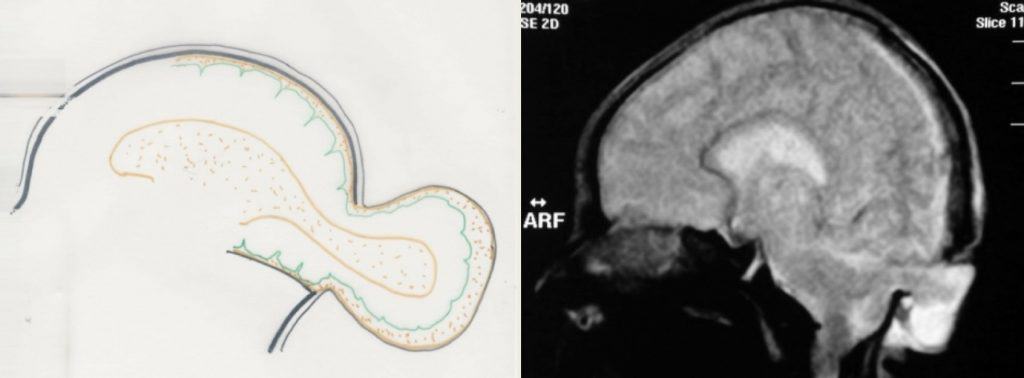
De acuerdo con la localización, los encefaloceles más frecuentes pueden ser:
A.- Occipital. – El niño recién nacido muestra una tumoración blanda a nivel occipital, con alteraciones cutáneas sobreañadidas. Se puede asociar con hidrocefalia.
B.- Frontobasales.- A nivel nasoetmoidal, esfenoidal…Su detección es mas tardía y suelen manifestarse como una tumoración en la raíz de la nariz o dentro de las fosas nasales.
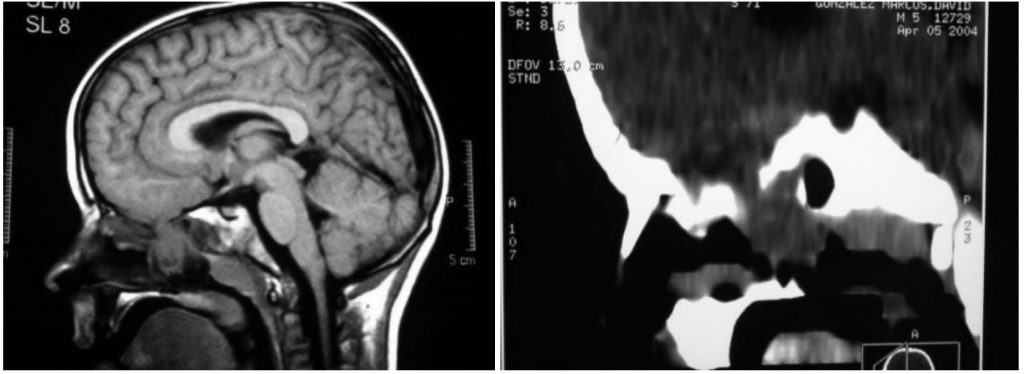
DIAGNOSTICO
Por la exploración clínica, Rx de cráneo para ver el defecto y, sobre todo, TAC o RNM para ver la localización, forma y contenido del tejido cerebral herniado
TRATAMIENTO
El tratamiento quirúrgico pretende reponer cada estructura en su compartimiento, si es posible. Es importante obtener siempre el cierre de la duramadre, aunque el defecto óseo puede esperar y ser reparado cuando el niño sea más mayor.
En casos de meningoceles solamente, se puede conseguir un desarrollo intelectual normal hasta en un 80-90% de los casos. Si se trata de un encefalomeningocele, el pronóstico es más sombrío, disminuyendo este porcentaje a un 20-40 %.
IX.- HOLOPROSENCEFALIA
Malformación por alteración en la inducción ventral con anomalía en la génesis cefálica y facial. Es un defecto de línea media. Se produce en la 4ª-5ª semana de gestación
B.- ALTERACIONES EN LA PROLIFERACION NEURONAL
I.- MICROCEFALIAS Y MEGALENCEFALIA
Son alteraciones que se producen hacia el 2º- 4º mes del desarrollo. En el primer caso hay una reducción en el número de neuronas y en el segundo un aumento. Se deben a factores familiares y prenatales.
La microcefalia, suele asociarse a irradiaciones o exposición a toxinas. La microcefalia vera consiste en un cerebro pequeño (microencefalia), con perímetro cefálico con un percentil menor del 10%, tienen retraso intelectual y no suelen tener crisis epilépticas.
Por el contrario en la megalencefalia uno de los hemisferios cerebrales aumenta de forma anormal su tamaño y el niño presenta una epilepsia grave o catastrófica, que precisa tratamiento quirúrgico (hemisferectomía o hemisferotomía) para controlar las crisis y evitar un grave retraso psicomotor.
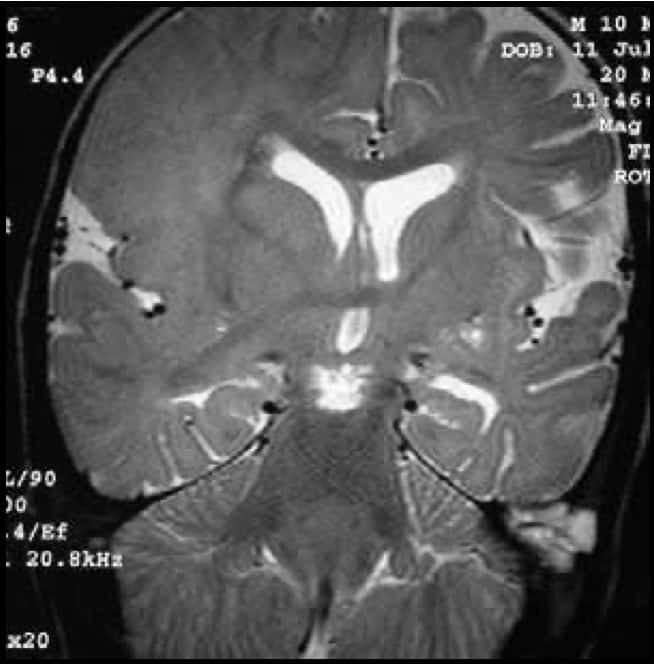
II.- ANENCEFALIA E HIDRANENCEFALIA
Es la falta de desarrollo de los hemisferios cerebrales. Sólo existen las estructuras de línea media. El resto esta ocupado por líquido cefalorraquídeo
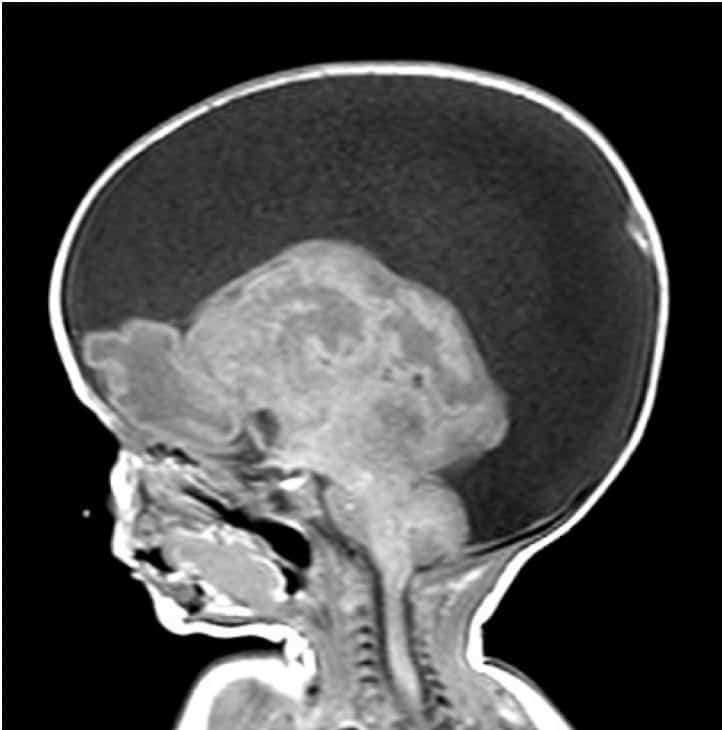
C.- ALTERACIONES EN LA MIGRACION:
Suceden entre las semanas 26 y 28 y consisten en el desarrollo anormal del patrón de circunvoluciones del manto cerebral. Prácticamente todas van a cursar con epilepsias graves fármaco-resistentes:
1.- LISENCEFALIA:
No se forman circunvoluciones cerebrales (agiria), siendo el córtex cerebral plano.
2.- PAQUIGIRIA:
Formación de circunvoluciones anormalmente gruesas, en las que hay un defecto de la configuración normal en seis capas.
3.- ESQUISENCEFALIA:
hay una hendidura anormal que une los ventrículos y la corteza, que puede ser uni o bilateral.
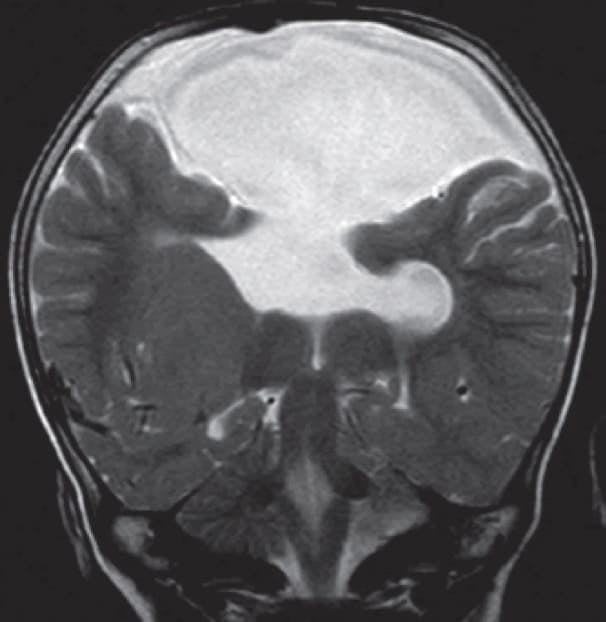
4.- AGENESIA DEL CUERPO CALLOSO:
Los axones que deberían haber cruzado a través del cuerpo calloso se disponen formando haces longitudinales en la cara interna de los hemisferios cerebrales. El sistema ventricular esta dilatado y a veces hay quistes interhemisféricos. Puede asociarse con otras malformaciones.
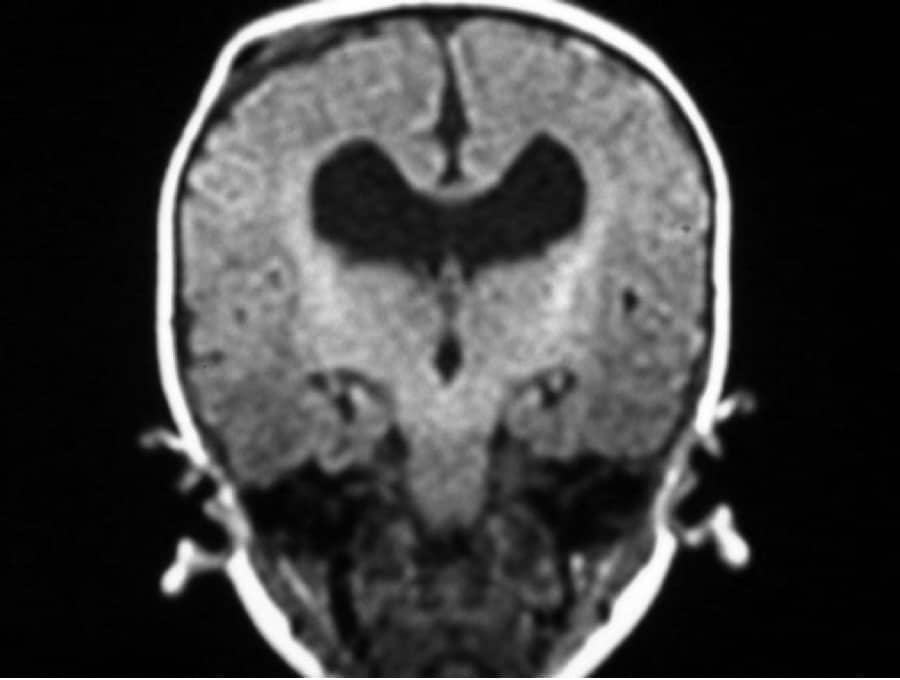
5.- HETEROTOPIAS CEREBRALES:
Por migración neuronal deficiente y acumulación de neuronas aberrantes en cualquier punto entre el epéndimo ventricular y la corteza cerebral.
6.- DISPLASIAS CORTICALES:
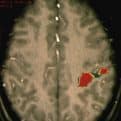
Alteraciones más focales del proceso de migración neuronal. A diferencia de todas las anteriores, la epilepsia que producen puede tener mejor pronóstico, dado que la cirugía de localización y exéresis del foco displásico es posible hoy día y obtiene resultados cada vez más alentadores. (Fig 22)
PARTE II – MALFORMACIONES CONGENITAS RAQUI-MEDULARES
El concepto, etiología y patogenia son similares a lo referido anteriormente. Incluso a veces coexisten en el mismo individuo ambos tipos de malformaciones.
CLASIFICACION
La clasificación es compleja. Por lo que, en aras de un mejor entendimiento, dividiremos este capítulo en dos grandes apartados: Malformaciones a nivel de la unión cráneo-cervical y malformaciones a nivel de la unión lumbosacra, por ser las dos regiones donde se producen con mayor frecuencia alteraciones durante el periodo de formación del SNC y sus cubiertas:
A.- MALFORMACIONES A NIVEL DE LA UNIÓN CRÁNEO-CERVICAL
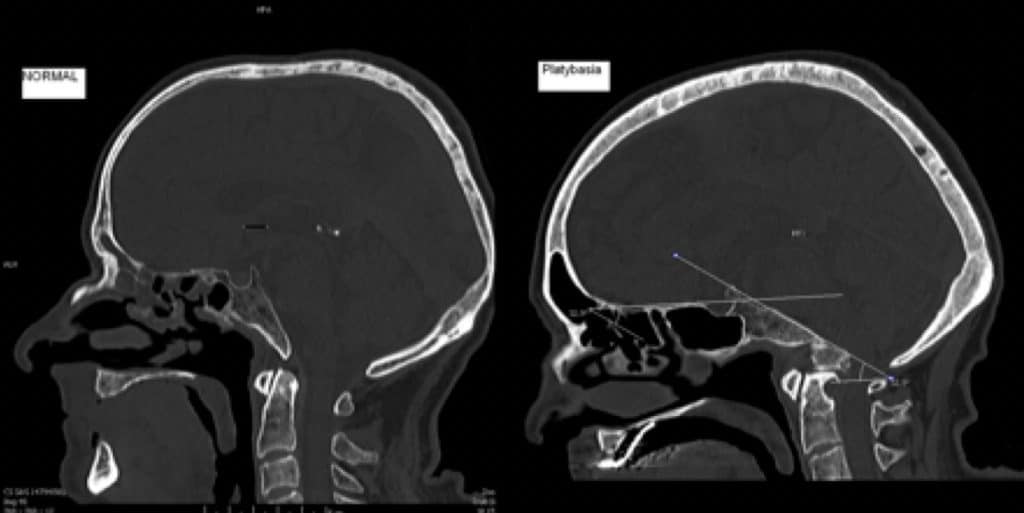
I.- PLATIBASIA
Consiste en un aplanamiento de la base del cráneo, con una apertura o aumento anómalo del llamado «ángulo basal» o de Mac Rae. Este ángulo lo forma la intersección en el centro de la silla turca de dos líneas dibujadas sobre la base craneal: una desde el nasion y otra desde el borde anterior del agujero magno. La apertura normal de este ángulo es de 120º a 145º. Se considera que existe una platibasia si es mayor de esta medida.
La platibasia no tiene repercusión en sí misma sobre el encéfalo, ni requiere tratamiento. Pero es un índice indirecto de posible presencia de malformaciones del SNC. Se debe completar el estudio con resonancia y/o TAC
II.- IMPRESION BASILAR
Es la malformación más frecuente de la charnela occípito-cervical y consiste en el hundimiento del cráneo sobre la columna cervical. La base del cráneo está descendida con respecto al límite superior de la odontoides. Puede ser congénita (Osteogénesis Imperfecta, por ejemplo) o adquirida (Enfermedad de Paget o lesiones tumorales).
Al “ceder” la región occipital, la odontoides tiene tendencia a introducirse en el agujero magno, generando un síndrome de compresión medular alta.
CLINICA
Dolor a nivel occípito-cervical y progresivo síndrome de compresión medular alta.

DIAGNOSTICO
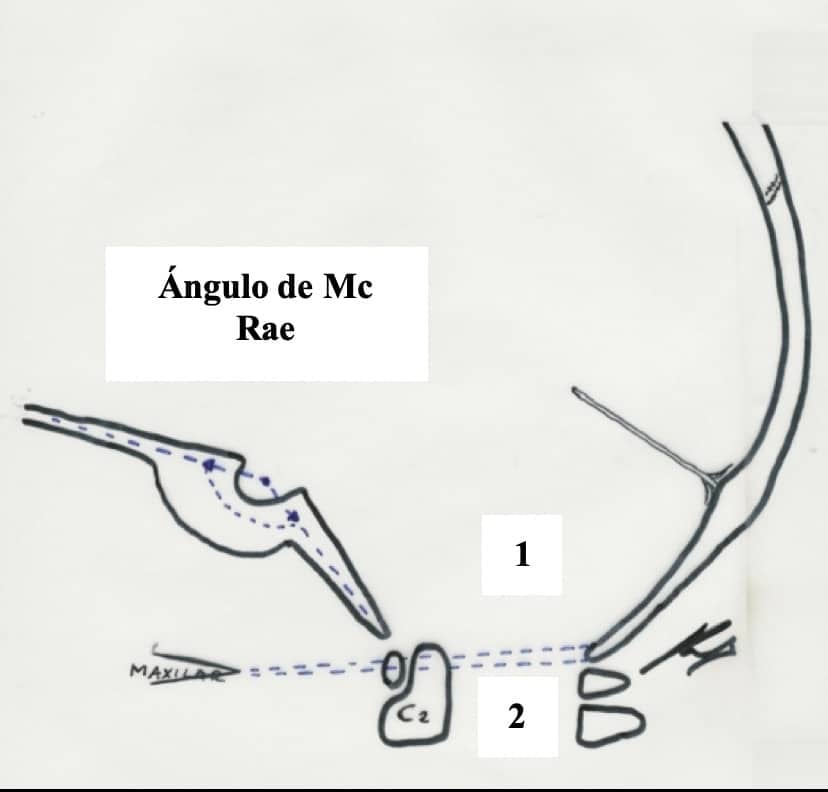
Con una simple Rx de cráneo podemos efectuar el diagnóstico, dibujando una serie de líneas ya clásicas (Figs. 26, 27):
– Linea de Chamberlain : desde el paladar duro al borde posterior del agujero occipital.(1)
– Linea de Mc Gregor: desde paladar duro a la parte mas baja del hueso occipital. Se considera impresión basilar si la odontoides sobrepasa mas de 5-7 mm. (2)
– Linea bimastoidea de Fischgold.
– Línea bidigástrica. Al igual que la anterior, se mide en una proyección A-P de cráneo. Si la punta de la mastoides sobrepasa ya esta línea estamos ante una impresión basilar severa.
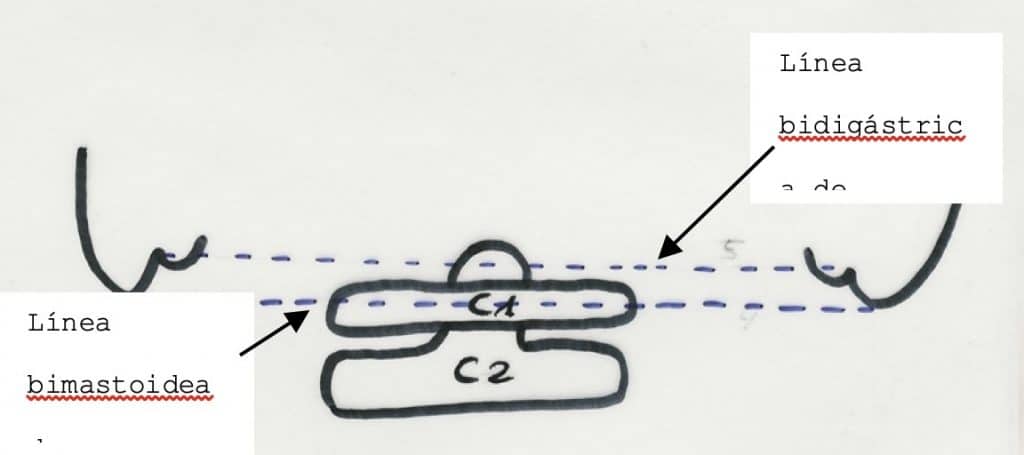
La impresión basilar suele ser indicativo de que existen otras malformaciones del SNC a este nivel, entre la que destaca el llamado Chiari I y la siringomielia, que aumentan aún más las posibilidades de compresión y afectación medular respectivamente. Por tanto, el diagnóstico correcto ha de establecerse completando el estudio con RM craneal y cérvico-dorsal, para visualizar el resto de las malformaciones.
TRATAMIENTO
Por sí sola es posible que la impresión basilar no precise tratamiento quirúrgico. Pero su asociación con las otras malformaciones indica la necesidad de realizar una cirugía descompresiva, con craniectomía occipital descompresiva (extirpación de la concha occipital y apertura del agujero magno), seguida o no de una estabilización de la charnela occípito-cervical. A veces es preciso, además, realizar una descompresión anterior, con resección de la odontoides introducida en el agujero magno.
III.- MALFORMACIONES DE LA CHARNELA OCCIPITO-CERVICAL
A este nivel hay un gran número y variedad de alteraciones óseas, fundamentalmente por anomalías en el diseño de las 2 primeras vértebras cervicales. Todas pueden llegar a favorecer una clínica de compresión medular alta. Podemos destacar las siguientes:
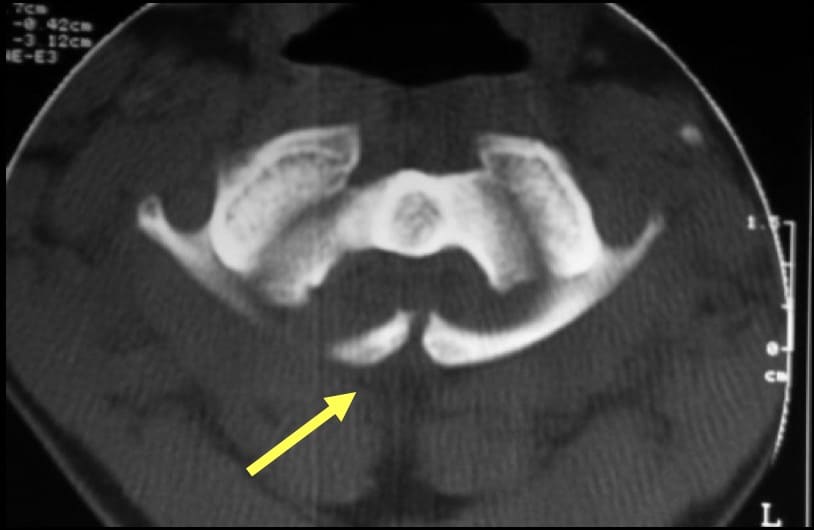
1.- MALFORMACIONES DEL ATLAS.
Consistentes en displasias, espina bífida y defectos de osificación. Pero la más frecuente es la occipitalización del atlas, en la que el atlas se fusiona al occipital, de forma total o parcial.
2.- MALFORMACIONES DE LA APOFISIS ODONTOIDES
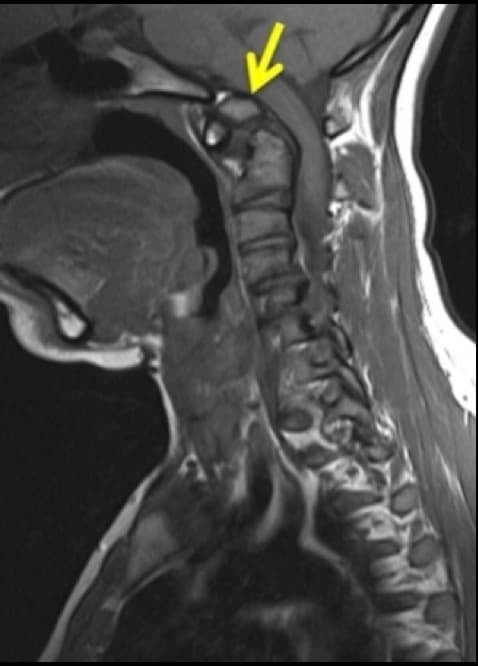
Destacaremos la existencia del osículo terminal de Bergmann, que consiste en la persistencia del núcleo de osificación apical de la odontoides (Os Odontoideo); la odontoides móvil de Bevan (defecto en la unión de la odontoides con el cuerpo del axis); así como la existencia de odontoides hipoplásicas. Ninguna de ellas suele requerir tratamiento quirúrgico.
Pero es conveniente su conocimiento, dado que, tras traumatismos graves, podría ocasionarse una desestabilización de la unión atlo-axoidea, con luxación y riesgo de compromiso bulbo-medular.
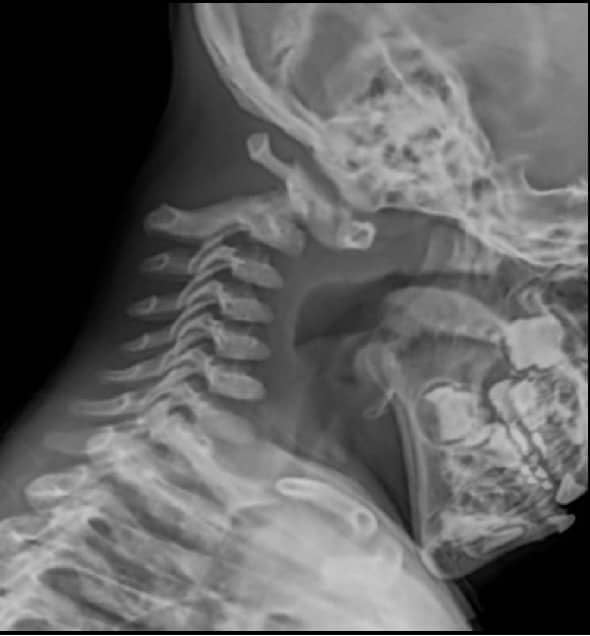
3.- LUXACION ATLO-AXOIDEA
Se presenta en casos de anomalía del ligamento transverso, que fija el atlas a la odontoides. En los niños la separación entre la odontoides y la parte posterior del arco anterior del atlas puede ser normal hasta 5 mm, aunque en los adultos es inferior a 2 mm. Cuando estas distancias son superiores, hay luxación atlo-axoidea.
Ésta se acentúa con los movimientos de flexión, llegando a producir compresión de la unión bulbo-medular, con síndrome piramidal e incluso parada respiratoria.
El tratamiento es quirúrgico y consiste en la fijación de los arcos posteriores del atlas y axis. En algunas ocasiones es preciso ampliar la fijación a la escama occipital. Hoy día se realiza con sistemas de placas y alambres de titanio, expresamente diseñados para esta región
IV.- SINDROME DE KLIPPEL-FEIL
Se trata de una fusión anómala de dos o más vértebras, debido a un fallo en el proceso de segmentación vertebral. Se da con una frecuencia aproximada de 1/50.000 personas y es más frecuente a nivel de C2-C3 o en el resto de las vértebras cervicales, siendo excepcional a nivel torácico o lumbar.
La clínica consiste en apariencia de cuello corto e inicio precoz de síntomas y signos de espondiloartrosis cervical (dolor, radiculopatía y/o mielopatía).
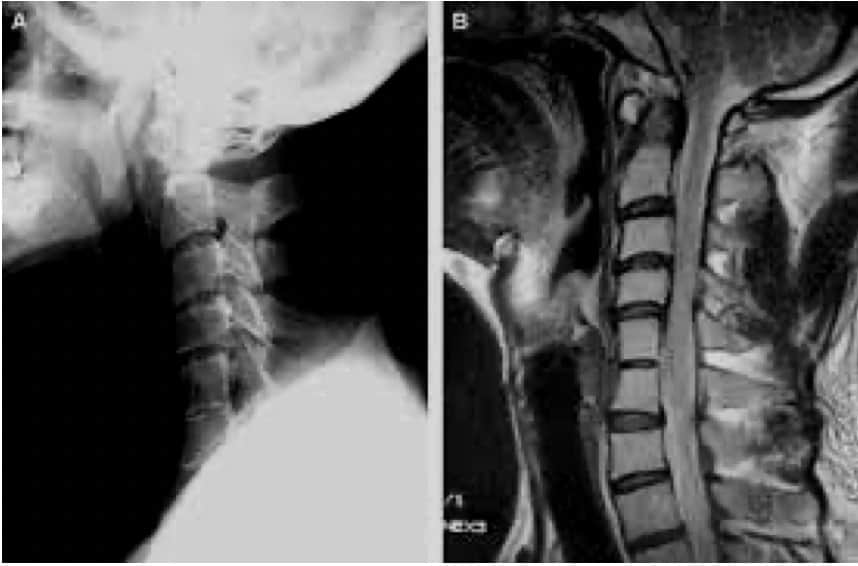
V.- MALFORMACION DE ARNOLD-CHIARI
Consiste en una elongación e introducción del cerebelo (amígdalas cerebelosas) en el agujero magno y canal vertebral.
Chiari las clasificó en 4 tipos:
– Tipo I.- Sólo están descendidas las amígdalas cerebelosas.
– Tipo II.- Descienden las amígdalas más la parte inferior del vermis e incluso la parte inferior del IV ventrículo. Se asocia siempre con espina bífida abierta e hidrocefalia. aparece en niños.
– Tipo III.- Todo el cerebelo está descendido y existe encefalocele occipital.
– Tipo IV.- Hipoplasia del cerebelo.
En la actualidad se distinguen solamente 2 entidades:
o Chiari tipo I.- Se presenta clínicamente en la edad adulta.
o Chiari tipo II.- Se presenta en niños asociado con espina bífida e hidrocefalia.
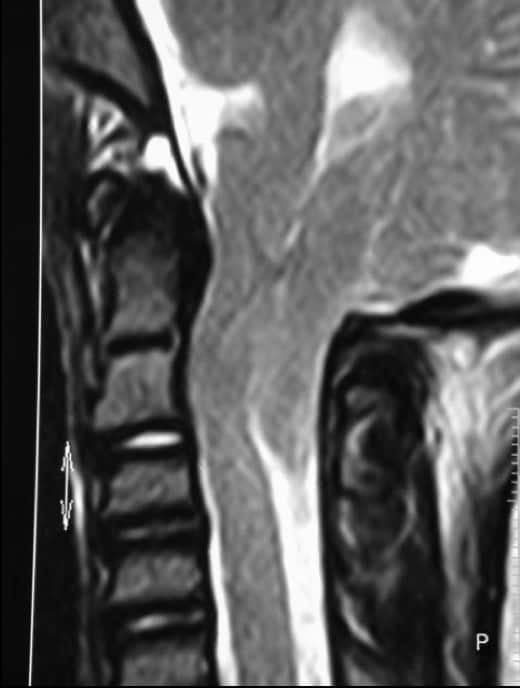
Es frecuente que ambas entidades se asocien con otras malformaciones neurales, entre las que destacan la Siringomielia y la Siringobulbia; así como malformaciones óseas ya descritas (Impresión basilar, occipitalización del atlas, Klippel-Feil).
PATOGENIA
Para explicar la aparición del Chiari I y II se plantean varias teorías:
– Existencia de tracción a nivel del mielomeningocele
– Hidrodinámica de Gardner: el retraso en la apertura de los agujeros de Luschka y Magendie hace que la presión del LCR desplace el cerebelo hacia el canal vertebral (fig. 33).
CLINICA
Por sí solas estas anomalías producen un síndrome de compresión a nivel del agujero magno (unión bulbo-medular), agudizado si además existe una impresión basilar. La aparición de los síntomas suele ser en la edad adulta. Aparte hay que considerar otras anomalías con las que se puede asociar, como Hidrocefalia o Siringomielia.
TRATAMIENTO
El mismo descrito en la impresión basilar, con la salvedad de que, en ocasiones, la descompresión (craniectomía suboccipital) ha de incluir los arcos posteriores del atlas y axis, dado el descenso de las amígdalas cerebelosas hasta este nivel (Fig. 32). Debido a que la presencia de las amígdalas cerebelosas han hecho desaparecer la cisterna magna, ésta se reconstruye abriendo la duramadre ampliamente y colocando una plastia que permita ampliar el contenido de la fosa posterior.
VI.- SIRINGOMIELIA
Es una dilatación congénita del canal ependimario. Se denomina también, con mayor propiedad, hidromielia.
PATOGENIA
La ya referida Teoría Hidrodinámica de GADNER intenta explicar con un solo proceso patogénico todas las alteraciones congénitas. Aunque no está demostrada, tiene la virtud de explicar de una forma elegante todos los procesos y hacer fácil su compresión.
En el caso de la siringomielia, la inadecuada permeabilidad de los agujeros que comunican el IV ventrículo con los espacios subaracnoideos hace que el LCR intente salir por el obex hacia el conducto ependimario. Éste se va a dilatar como cuando inflamos un globo alargado: dependiendo de la elasticidad de las paredes se dilata en puntos diferentes y, a partir de aquí, manteniendo la misma presión la dilatación va aumentando. Aunque incrementáramos la presión, es muy difícil que se dilaten otras zonas.
En la siringomielia, las cavidades más frecuentes están situadas en región cervical y dorsal. Ocasionalmente también se extiende hacia arriba (siringobulbia). Se asocia prácticamente siempre con un Chiari I, impresión basilar y/o alteraciones de la charnela occipito-cervical.
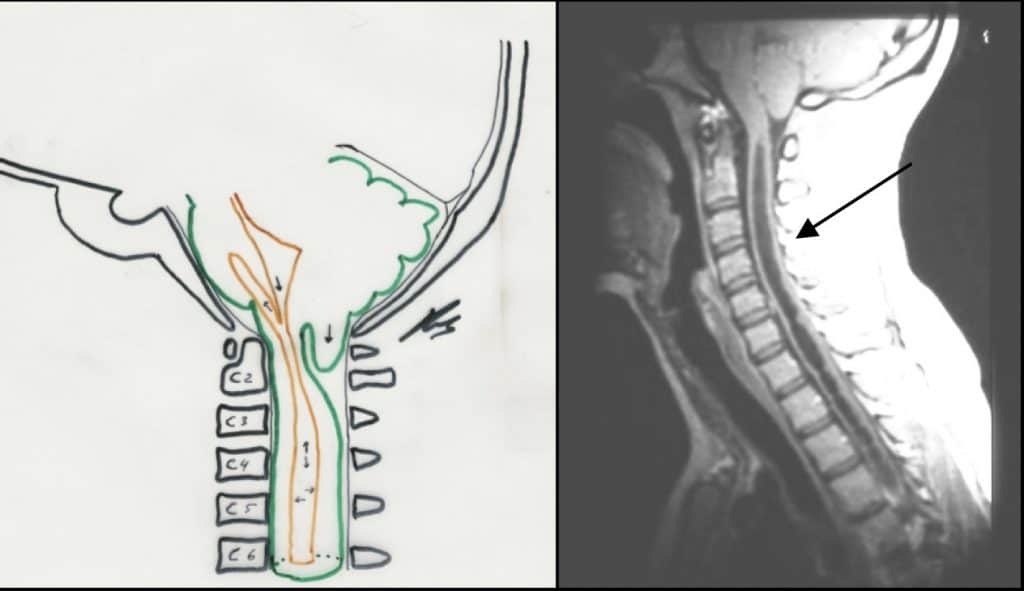
CLINICA
Suele presentarse a partir de la tercera década, con un síndrome característico, disociación siringomiélica, consistente en afectación de la sensibilidad termo-algésica (nota menos el calor y no tiene dolor, llegando incluso a producirse quemaduras sin darse cuenta), con preservación de la sensibilidad táctil que discurre por los cordones posteriores medulares. Estas alteraciones se sitúan a nivel del quiste siringomiélico (extremidades superiores, en el caso más frecuente de afectación medular cervical).
A medida que el cuadro progresa, aparecen déficits motores en extremidades superiores, con atrofias musculares por lesión de motoneurona anterior; alteraciones tróficas y vasomotoras (manos frías, cianóticas y húmedas) y síntomas de afectación de vías largas (paraparesia espástica). Se asocia frecuentemente escoliosis, probablemente por debilidad de la musculatura paravertebral.
Si existe además siringobulbia, se sobreañaden síntomas de afectación de pares craneales bajos (IX a XII).
Hay que saber que esta malformación, una vez que inicia los síntomas, tiene una evolución lenta y progresiva.
DIAGNOSTICO
La clínica es característica por el cuadro de disociación siringomiélica, de fácil exploración clínica.
En las Rx simples de cráneo podemos ver si existen anomalías de charnela o impresión basilar asociadas.
La RM es la prueba diagnóstica de elección pues visualiza muy claramente la lesión siringomiélica en toda su extensión y las posibles lesiones acompañantes, como Chiari I, hidrocefalia, etc.
La RM es además una exploración imprescindible para el seguimiento clínico de estos pacientes, intervenidos quirúrgicamente o no.
TRATAMIENTO
Hay una cierta controversia sobre cuál es el mejor método quirúrgico. Una forma de tratarla es introducir un catéter fino en la cavidad (Siringostomía), que se deja drenando en el espacio subaracnoideo perimedular. Tiene el inconveniente de que hay que perforar la médula para introducir el catéter (aumentando la lesión de cordones posteriores) y que los catéteres se acaban obstruyendo.
Otro tipo de abordaje es la craniectomía de fosa posterior, abrir el IV ventrículo y colocar un pequeño trozo de músculo en el obex, para impedir que la pulsación del LCR mantenga la cavidad a tensión. Esta intervención ha sido propuesta por Gardner (Fig. 33). En la actualidad se está viendo que no es necesario cerrar el obex y que la amplia craniectomía, con reconstrucción de la cisterna magna (como ya hemos referido en el apartado del Chiari I), mejora la siringomielia. Esta intervención, además, solventa el problema del Chiari I con el que se asocia casi ineludiblemente la siringomielia.
Hay un tercer tipo de intervención, propuesta también por Gardner, consistente en seccionar el filum terminale, a nivel lumbosacro, para que el LCR del canal ependimario drene en el fondo de saco dural. En el filum terminale se encuentra en ocasiones una pequeña dilatación del canal ependimario (llamada V ventrículo). Si el canal ependimario es permeable en toda la extensión (aunque no se pueda visualizar en la RM), esta intervención sería una siringostomía más fisiológica. Esta intervención está en desuso.
La intervención quirúrgica presenta unos resultados aproximados de mejoría en, al menos, un 50% de los casos y detención de la enfermedad en un porcentaje algo menor, aunque existen unas probabilidades de hasta un 20% de empeoramiento del cuadro clínico.
B.- MALFORMACIONES A NIVEL DE LA UNIÓN LUMBOSACRA
I.- ANOMALIAS DE TRANSICION
Es muy frecuente encontrar anomalías en la transición lumbo-sacra, con lumbarización de la primera vértebra sacra (parece que hay 6 vértebras lumbares) o sacralización de la 5ª vértebra lumbar (apariencia de un sacro anormalmente grande).
No tienen significación clínica y pueden ser tenidas, por su frecuencia, casi como variantes de la normalidad. Pero, en ocasiones, esta anomalía provoca alteraciones dinámicas que van a repercutir en la funcionalidad del disco intervertebral interpuesto y ser ocasión de degeneraciones precoces de éste o incluso su herniación.
Ante estos hallazgos (clínica de discopatía lumbosacra y anomalías de transición) hay que realizar, aparte de los estudios habituales de RM, estudios dinámicos de columna lumbar para observar si existe, además de una hernia discal, una inestabilidad vertebral. En estos casos, al mismo tiempo que la microdiscectomía convencional, es posible que nos tengamos que plantear fijar ambas vértebras inestables con los sistemas actuales de tornillos transpediculares.
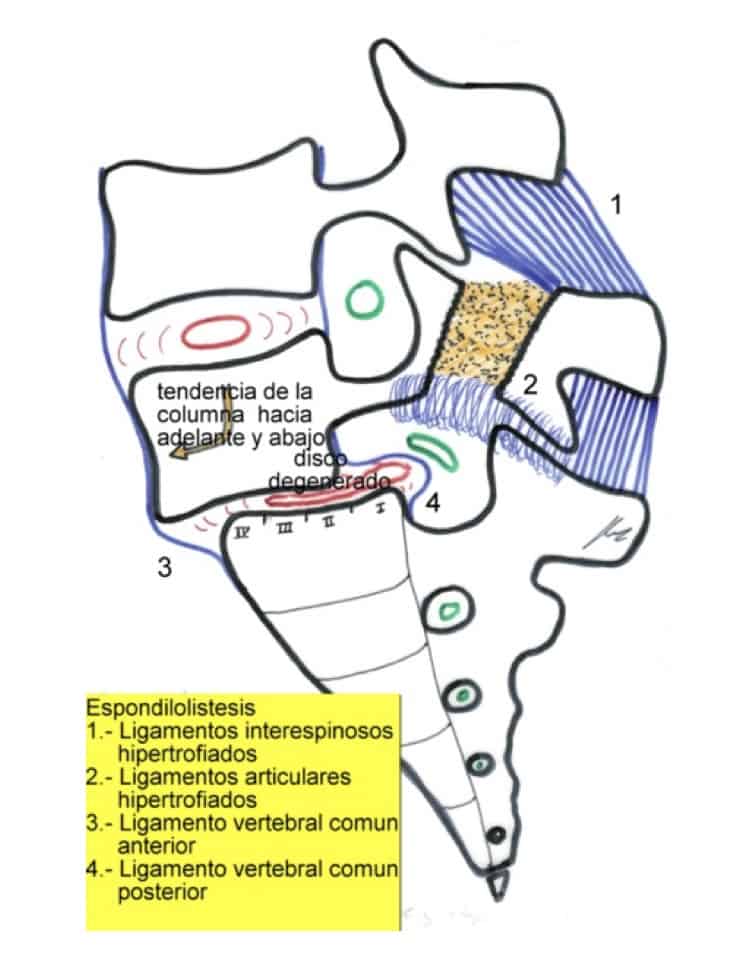
II.- ESPONDILOLISIS-ESPONDILOLISTESIS
Consiste en un defecto de osificación de la pars articularis de una vértebra (espondilolisis). Esta solución de continuidad bilateral en el arco vertebral provoca la incapacidad de fijar una vértebra sobre la otra, por lo que se va ocasionando un desplazamiento hacia delante del cuerpo vertebral superior sobre el inferior (espondilolistesis).
La localización más frecuente es a nivel de L4-L5 o L5-S1. Hay 4 grados, dependiendo del porcentaje de distancia antero-posterior del cuerpo vertebral que hay de desplazamiento (25, 50, 75 y 100%).
CLÍNICA
Se inicia con dolor lumbar y posteriormente aparece radiculopatía Acaba produciéndose una estenosis del canal, con compresión de la cola de caballo y claudicación intermitente al caminar, para finalizar en una paraparesia progresiva.
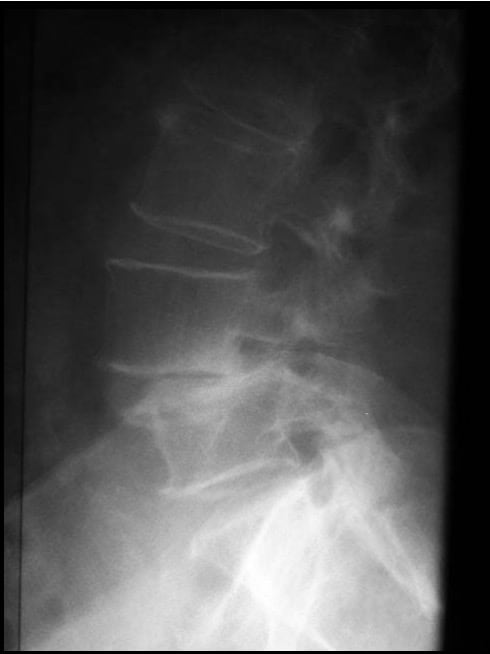
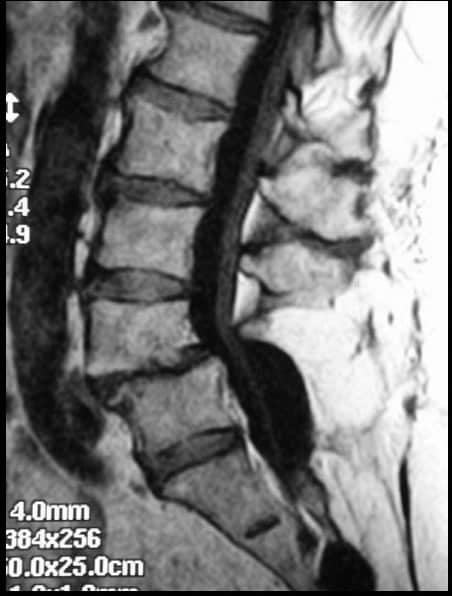
DIAGNÓSTICO
Es radiológico (en la Rx lateral se aprecia la listesis (Fig 36) y en la Rx oblicua se confirma la imagen de lisis). Se completa el estudio con TAC y RM. (Fig 37)
TRATAMIENTO
Ha de realizarse en la mayoría de los casos una descompresión posterior (laminectomía), seguida de la fijación mediante tornillos pediculares, completando la artrodesis con hueso de cresta iliaca del propio paciente. En casos muy acentuados esta artrodesis ha de ser más compleja (intersomática o incluso con fijación 360 grados).
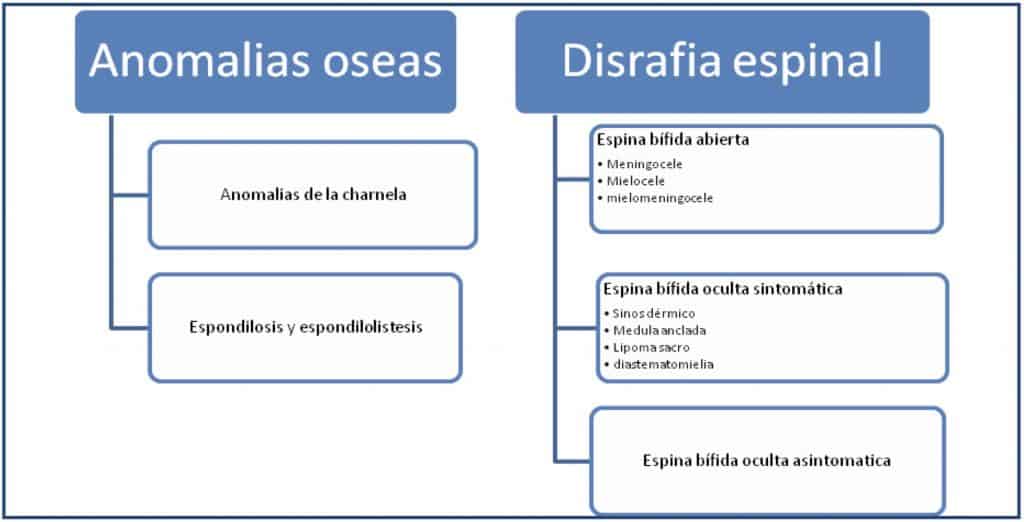
III.- DISRAFIA ESPINAL
Este apartado es complejo de clasificar y confuso en su terminología. El concepto de disrafia implica un defecto de la fusión del tubo neural y de sus estructuras cutáneas, musculares y óseas adyacentes. Otra forma de abordarlo terminológicamente es el de espina bífida, que implica la falta de soldadura del arco posterior vertebral y la apariencia bífida a la palpación de la parte posterior de la vértebra.
Si se conjugan ambas terminología, se podría hacer la clasificación referida anteriormente y que volvemos a reproducir:
Disrafia espinal.- Como término que engloba a todos los siguientes procesos:
o Espina bífida oculta asintomática
o Espina bífida oculta sintomática:
– Sinus dérmico
– Médula anclada
– Lipoma sacro
– Diastematomielia
– …
o Espina bífida abierta
– Meningocele
– Mielocele
– Mielomeningocele
Similar al caso del encefalocele, en este grupo de malformaciones el fallo del cierre del tubo neural se encuentra, sin embargo, a nivel del neuroporo posterior o caudal. Esto induce alteraciones neurales y mesenquimatosas. Se produce a lo largo de los primeros 3 meses de gestación.
A partir de estos mesas, lo normal es que comienza a crecer más la columna que la médula, ascendiendo el extremo distal de ésta hasta el nivel de la 1ª-2ª vértebra lumbar, ya que en el extremo proximal la médula está fija por el tronco del encéfalo. Si hay un fallo en el cierre del neuroporo posterior, la médula queda anclada a éste.
1.- ESPINA BIFIDA ABIERTA
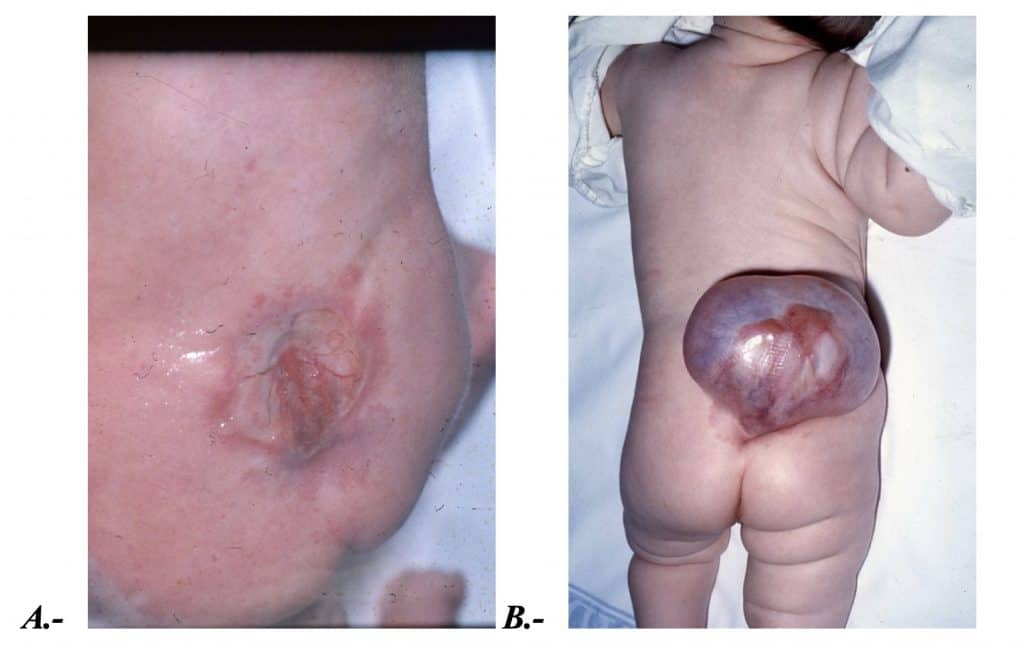
También denominada espina bífida quística o mielodisplasia. Se caracteriza porque hay un defecto de cierre en la piel, arco posterior vertebral y médula. Puede localizarse a nivel tóraco-lumbar, lumbar o sacro. El déficit neurológico dependerá del nivel y de la gravedad de la falta de cierre del tubo neural.
CLASIFICACION
MIELOCELE o MIELOSQUISIS.- La placa neural está abierta y expuesta a la intemperie. El LCR sale espontáneamente y hay un déficit neurológico completo por debajo de la lesión. El fallo en el cierre sucede antes del día 28.
MIELOMENINGOCELE.- El tubo neural está casi cerrado, pero hay una zona quística epitelizada en comunicación con el espacio subaracnoideo, no sale LCR y se produce menor alteración neurológica por debajo de la lesión. En ocasiones las raíces de la cola de caballo sobrenadan en la cavidad quística. Sucede después del día 28 de gestación.
MENINGOCELE.- Hay sólo raquisquisis (falta de cierre del arco posterior vertebral). El contenido es únicamente LCR recubierto por duramadre y por la piel, manteniendo la comunicación con el espacio subaracnoideo. La médula en estos casos no presenta alteraciones, por lo que la lesión neurológica es mínima si existe.
Todas estas malformaciones se asocian con descenso de amígdalas cerebelosas (Chiari II). Su incidencia puede llegar al 2‰ de los nacimientos y predomina en sexo femenino.
PATOGENIA
Hay varias teorías para explicar esta malformación. Van desde suponer la detención en el desarrollo y cierre del tubo neural por excesivo crecimiento del tejido nervioso, que provoca la eversión de los pliegues neurales, impidiendo el cierre. O la ya referida Teoría Hidrodinámica de Gadner: una vez cerrado el tubo neural, se vuelve a abrir por presión y acúmulo de LCR en el canal ependimario, debido al retraso en la apertura del IV ventrículo hacia cisterna magna.
CLINICA
La malformación es visible en el momento de nacer. Dependiendo de la altura en que se localice y del tipo de afectación del tubo neural, el recién nacido presentará una lesión motora y sensitiva más o menos completa en las extremidades inferiores junto con alteración del esfínter vesical y anal.
Se asocia con un descenso de amígdalas cerebelosas (Chiari II) y en un alto porcentaje de casos con hidrocefalia por estenosis de acueducto de Silvio.
DIAGNOSTICO
Se puede llevar a cabo durante el embarazo, midiendo la α-fetoproteina en el suero de la madre o en líquido amniótico. También se puede detectar mediante ecografía o RM. El diagnóstico precoz intrauterino plantea el problema ético de detención voluntaria del embarazo.
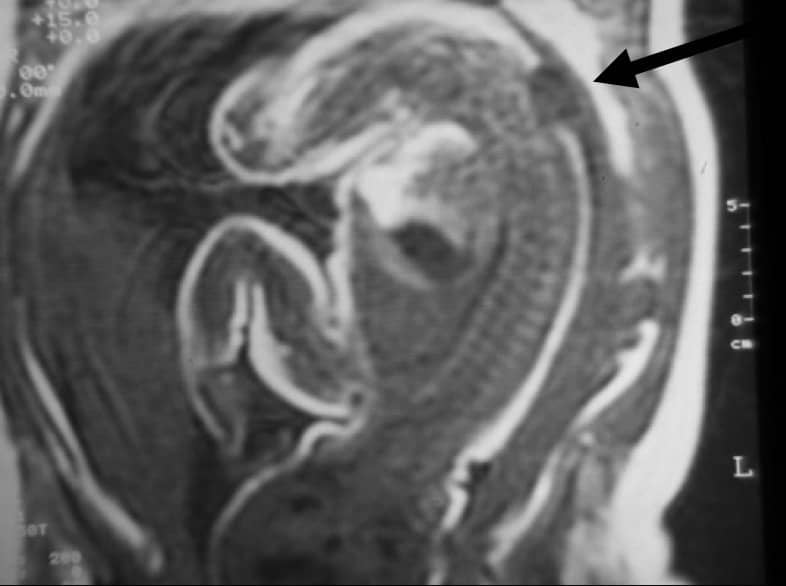
En el recién nacido es preciso realizar una exploración neurológica exhaustiva, para ver el nivel y las lesiones neurológicas existentes a nivel de extremidades inferiores y esfínteres.
La RM completa el proceso diagnóstico, que ha de incluir el estudio de craneal y de la charnela occípito-cervical, dada la frecuencia de malformaciones asociadas ya referidas.
TRATAMIENTO
Consiste en la reparación quirúrgica, reconstruyendo los planos de duramadre, músculo y piel, evitando la salida de LCR y cuidando de no lesionar la placa neural. Ha de ser realizado de forma urgente, sobre todo si hay salida espontánea de LCR.
Es muy frecuente que, una vez reparado, el niño desarrolle hidrocefalia que precise una derivación ventrículo-peritoneal.
Hay que tener en cuenta que estos niños han de ser tratados desde el principio con una perspectiva de abordar el problema en conjunto con un equipo multidisciplinario que incluya otros especialistas de rehabilitación, urología, ortopedia, psicología, etc. La finalidad es ir apoyando al niño en sus diferentes etapas de desarrollo, para obtener el mayor nivel de incorporación social posible.
2.- ESPINA BIFIDA OCULTA SINTOMÁTICA
En este apartado se incluyen una gran cantidad de procesos cuyo elemento común es la existencia de un defecto congénito del cierre del neuroporo posterior, que se manifiesta externamente con la presencia de espina bífida (claramente visible en la Rx de región lumbosacra) (Fig.41), pero que está acompañada de alteraciones más complejas, entre las que destacan:
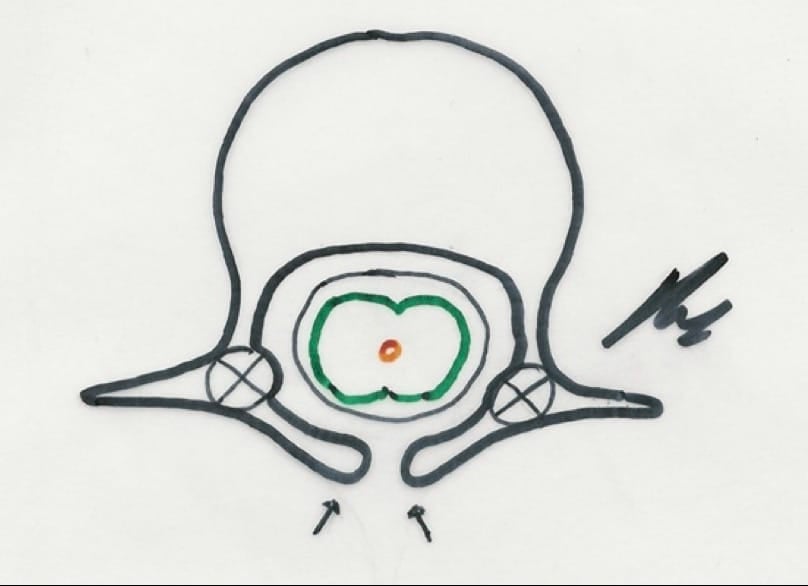
a.- Sinus dérmico.- Similar al referido a nivel occipital, con posibilidad de existencia de tumores de inclusión (dermoides o epidermoides). Hay que explorar la región lumbosacra ante un cuadro de meningitis de causa desconocida. El hallazgo de alteraciones cutáneas nos ha de llevar a la realización de Rx y RM de dicha región. El tratamiento debe incluir el trayecto y la posible tumoración existente.
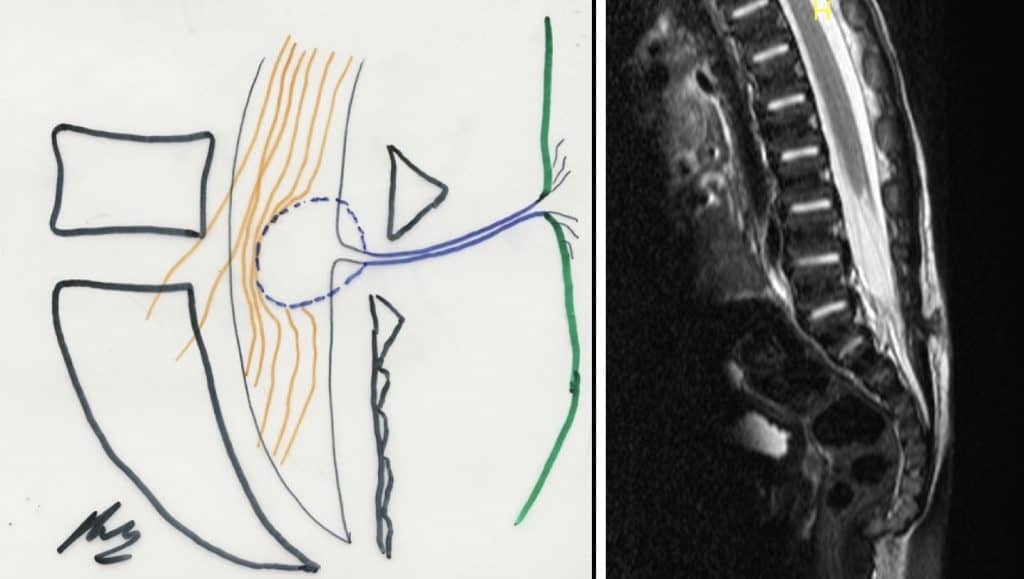
b.- Lipoma lumbosacro.- Existe un lipoma que puede llegar a envolver las raíces de la cola de caballo, haciendo muy difícil su extirpación completa. Por el contrario, a veces es sólo el filum terminale el que tiene un mayor contenido graso, que puede distinguirse muy bien en la RM.
c.- Teratoma sacro.- Lesión maligna e invasiva, que requiere intervenciones quirúrgicas muy agresivas
d.- Meningocele sacro anterior.- Que puede llegar a comprimir los órganos pélvicos.
e.- Quiste neuroentérico.- También a nivel sacro anterior, por persistencia de comunicación del tubo neural con el endodermo.
f.- Meningocele oculto intrasacro.- Difícil de distinguir en ocasiones de un quiste radicular intrasacro.
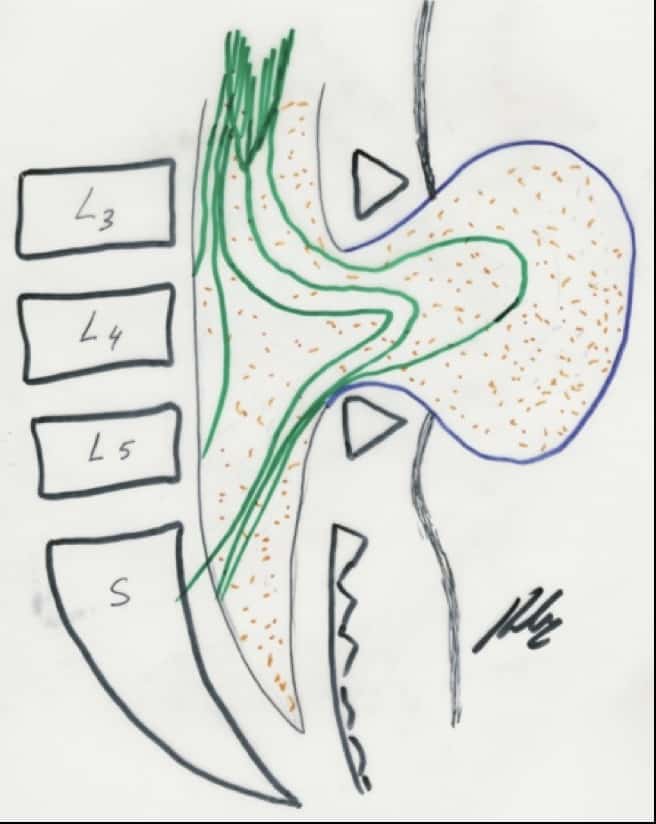
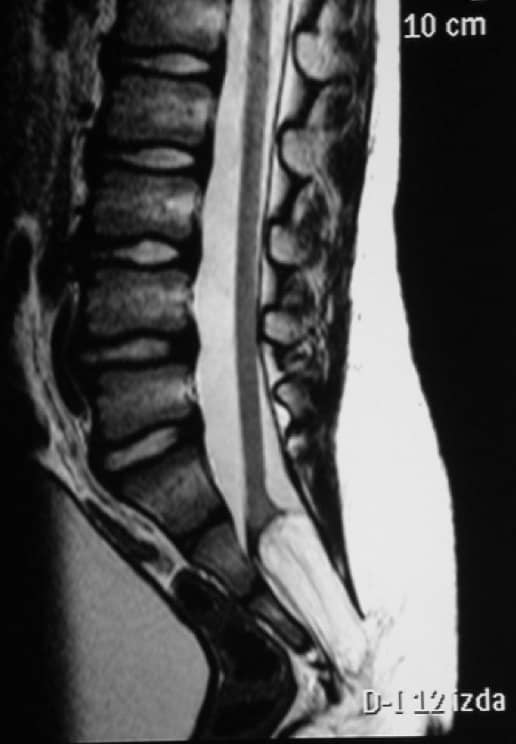
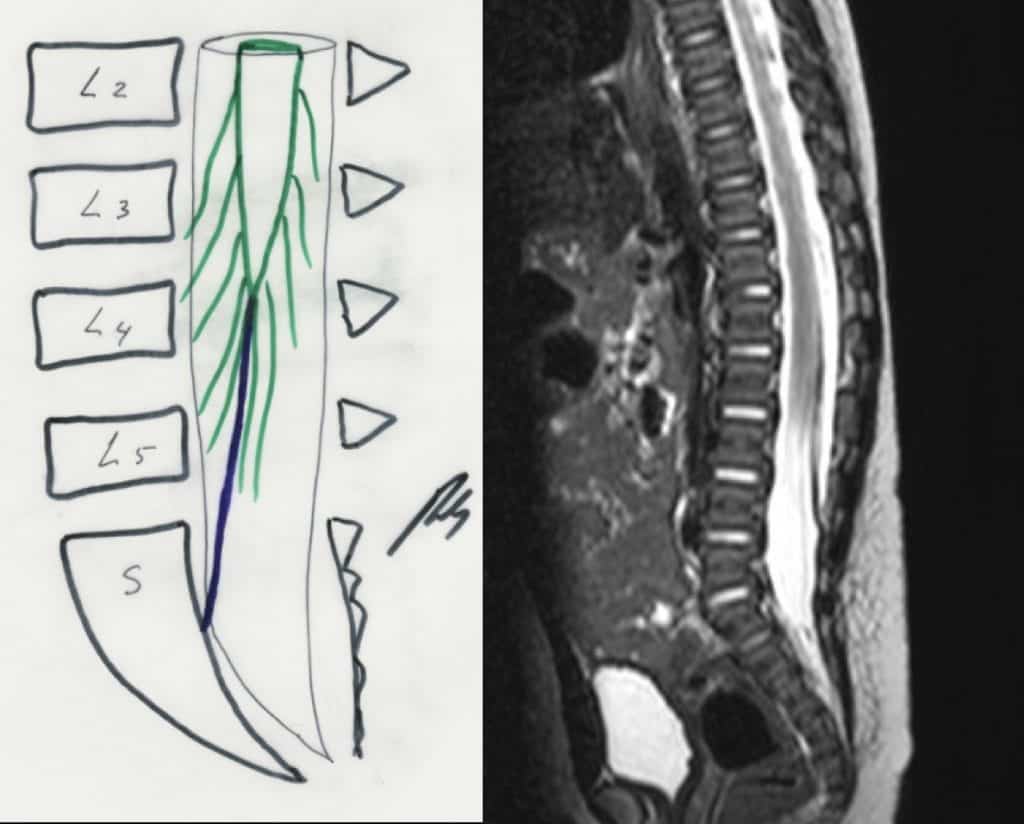
g.- Sindrome de médula anclada.- Hay un fallo en el extremo caudal de la médula, con un filum terminale más corto, por lo que queda el cono anclado. Provoca en la edad adulta molestias y dolores lumbares. Es más frecuente en mujeres. En la Rx lumbar vamos a visualizar una espina bífida a nivel de L5 o S1 y cómo el cono medular en la RM llega más debajo de L1-L2. Puede asociarse con los síndromes arriba descritos. El tratamiento es la laminectomía lumbar baja y sección del filum terminale (Fig. 44).
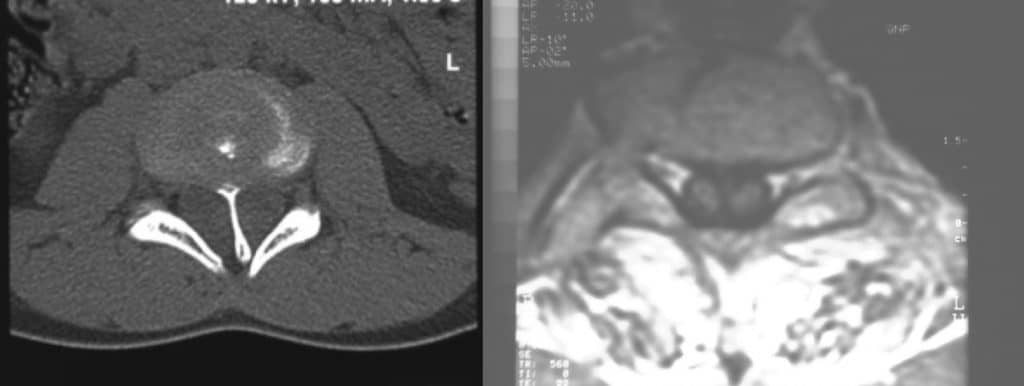
h.- Diastematomielia.- De localización dorsal y que consiste en un espolón óseo a nivel medio dentro del canal vertebral, en dirección antero-posterior, que divide la médula en dos mitades. Al crecer la columna, la médula queda anclada a este nivel, por lo que hay que realizar laminectomía y extirpar dicho espolón óseo (Fig 45).
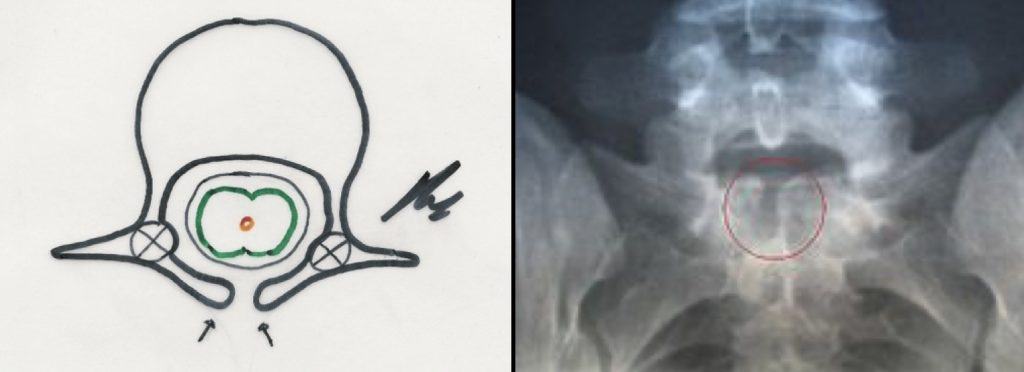
i.- Espina bifida anterior y posterior.- Puede ser sólo un hallazgo en los estudios de neuroimagen o asociarse con lesiones ya descritas.
3.- ESPINA BIFIDA OCULTA ASINTOMATICA
Es un fallo en la formación y cierre completo de láminas, por lo que no se forma la apófisis espinosa (Fig. 46). Por si sola, no supone afectación del tubo neural y no hay déficit neurológico.
Son asintomáticos y, por lo general, es un hallazgo casual en la Rx lumbosacra. Su localización más frecuente es en zonas de transición lumbosacro (L5 o S1) y más raro a otros niveles (cervical o torácica)
Si se descubre en la infancia hay que esperar hasta los 7 años, ya que puede ser un retraso en el cierre del canal. En los adultos los podemos encontrar en el 4,4% de la población, con mayor frecuencia a nivel S1 y en varones.
El diagnóstico precisa completarse con la RM, para descartar que existan otras alteraciones que la conviertan en una espina bífida sintomática. Dicha prueba se realizará dependiendo del grado de molestias clínicas que presente el paciente, si es adulto. En el caso de niños mayores, en los que persiste enuresis nocturna o tienen alteraciones en los pies (pie equino…), molestias lumbares, etc., es mandatoria esta exploración.
Debido a que supone una alteración a este nivel, con cierta frecuencia hay una afectación de la dinámica lumbosacra, con degeneración más precoz del disco intervertebral. En estos casos, los pacientes que presentan un cuadro de discopatía y espina bífida oculta han de ser estudiados, en cuanto a si presentan inestabilidad en los estudios dinámicos (Rx lateral de columna lumbosacra, en flexión y extensión máximas) y precisan un tratamiento quirúrgico más complejo, con microdiscectomía y fijación vertebral.
MALFORMACIONES DE SISTEMA NERVIOSO – CONCLUSIONES
- Mayor riesgo de recurrencia en madres con un hijo afecto (5%)
- Prevención: acido fólico antes de la concepción y durante las primeras 12 semanas de la gestación
- Diagnóstico prenatal: niveles de AFP en suero materno y líquido amniótico (en las primeras 18 semanas)
- En caso de niveles elevados de AFP: Ecografia de alta resolución (diagnostico de malformación antes de la semana 20)
- Gran parte no son susceptibles de tratamiento quirúrgico
- Es preciso un diagnóstico precoz con asesoramiento familiar y consejo genético en el caso de que existan malformaciones

buena noche dra. desde el punto analítico el tema de mal formaciones (MN) nos hace tomar conciencia respecto a ciertos cuidados en la etapa gestacional y planificación tomar en cuenta que tan importante es ver el consumo de bebidas alcohólicas, fármacos y de mas, también tomar en cuenta los controles prenatales para poder visualizar las formaciones del feto y ver si presenta algunas alteraciones en la etapa de desarrollo.
en el articulo nos da entender la variedad e malformaciones del sistema nerviosa que deberíamos tener mas en cuenta en el estudio de ellos.
Hola, me han hecho una Radiografía de la region Lumbosacro y como resultado tengo espina bífida oculta en S1. Desde que pasé mucho tiempo sentada presenté fuertes dolores en esa zona, el traumatólogo me dijo que eso no tiene nada que ver porque si fuese por la malformación que tengo, me hubiese dolido desde siempre y que me va a mandar a terapia porque mi columna está deviada, aunque eso no decia en los resultados, el traumatólogo no se veía profesional. Ahora aquí he leído que tiempo despues si puede presentar molestias. No sé pensar, a mí me dule justo… Leer más »