
Tema 4 – Enfermedades desmielinizantes
Sistema Nervioso Central y Sistema Nervioso Periférico
Dra. Paloma Pulido Rivas
Unidad de Neurocirugía RGS
ver CV completo
Introducción – Enfermedades desmielinizantes
Las enfermedades desmielinizantes son aquellas en las que el proceso patogénico principal está dirigido contra la mielina normal. Produciéndose una inflamación y/o destrucción selectiva de la mielina.
Es importante destacar que el origen de las vainas de mielina que envuelven los axones, provienen de distintos tipos de células gliales según se localicen en el sistema nervioso central (S.N.C) o periférico (S.N.P).
En el S.N.C las vainas de mielina son formadas por los OLIGODENDROCITOS. Cada oligodendrocito puede envolver varios axones de distintas neuronas (Figura 1A).
En el S.N.P, las vainas de mielina son formadas por las CÉLULAS DE SCHWANN. Cada célula de Schwann envuelve a un único axón. (Figura 1B).
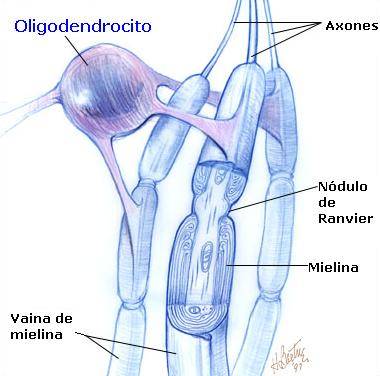
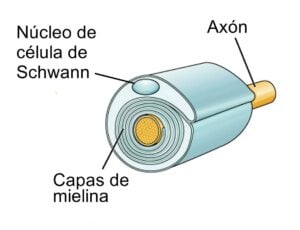
Las enfermedades que se van a estudiar dentro del grupo de enfermedades desmielinizantes, son las siguientes:
- ESCLEROSIS MÚLTIPLE
- ENCEFALITIS DESMIELINIZANTE AGUDA
- LEUCOENCEFALITIS MULTIFOCAL PROGRESIVA, relacionada con el SIDA
- SÍNDROME DE GUILLAIN-BARRÉ
- POLINEUROPATIA DESMIELINIZANTE CRÓNICA
1.- ESCLEROSIS MÚLTIPLE (EM)
Introducción
En cuanto a causas de discapacidad neurológica, y exceptuando a los traumatismos, ésta es la causa más frecuente de discapacidad neurológica en adultos jóvenes. Es la enfermedad por alteración de la mielina más frecuente en el sistema nervioso central (S.N.C.).
Se produce una destrucción selectiva de la mielina del sistema nervioso central.
Aunque la etiología es desconocida, se asocia su inicio con la aparición de un agente infeccioso, como un virus, que induciría una respuesta inmune, que conllevaría la destrucción de los oligodendrocitos, en un individuo genéticamente susceptible (el riesgo aumenta hasta en 20 veces si se tiene algún familiar afectado de dicha enfermedad).
Existe una clara diferencia en cuanto a la probabilidad de padecer esta enfermedad según el sexo. Las mujeres tienen de 2 a 3 veces más posibilidades de desarrollar EM que los hombres. En cuanto al rango de edad, la EM afecta principalmente a individuos de entre 20 y 40 años de edad.
Patológicamente, la EM se caracteriza por la presencia de lesiones del SNC, llamadas placas, que consisten en áreas de desmielinización de localización perivascular, que se localizan especialmente en el cuerpo calloso, nervios ópticos, tronco del encéfalo, cerebelo y médula espinal. Las lesiones antiguas presentan gliosis.
La incidencia, en España, de la EM es de 58 pacientes cada 100.000 habitantes por año.
Por definición, el sistema Nervioso Periférico nunca se ve afectado.
Formas Clínicas de la EM
(Nota: Se considera un brote a un déficit neurológico con una duración mayor de 24 horas)
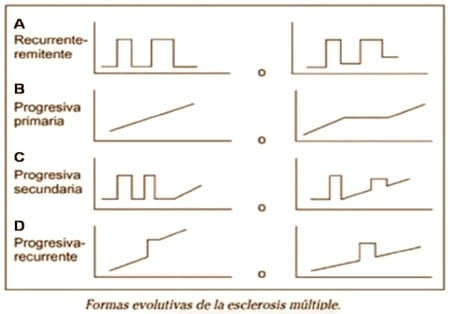
Se distinguen cuatro formas principales de EM:
- 1.- Forma Recurrente-Remitente (ver figura 1 A):
- Más frecuente en el inicio.
Se considera un brote a un déficit neurológico con una duración mayor de 24 horas.
Brotes de disfunción neurológica más o menos reversibles con remisión posterior.
Se considera que 2 brotes son diferentes si afectan a dos áreas distintas del S.N.C y con un intervalo de más de 30 días.
2.- Forma Progresiva Primaria (ver figura 1 B):
- Edad dependiente, y afecta con más frecuencia a pacientes de más edad; aquellos cuya enfermedad comienza durante o tras la quinta década de la vida
La sintomatología progresa desde el comienzo sin brotes ni remisiones.
Es la forma que es de más difícil diagnóstico y de peor pronóstico.
Representa aproximadamente el 10% de las formas evolutivas.
3.- Forma Progresiva Secundaria (ver figura 1 C):
- Tras un periodo de brotes, éstos disminuyen pero la discapacidad neurológica se sigue acumulando.
Es la forma evolutiva más frecuente, aproximadamente el 50%.
4.- Forma Progresiva Recurrente (ver figura 1 D):
- Deterioro progresivo desde el comienzo aparecen brotes.
Representa aproximadamente el 5% de las formas evolutivas.
Manifestaciones Clínicas de la EM
Motoras (aproximadamente en el 35% de los pacientes)
Con el curso de la enfermedad, los pacientes muestran un patrón característico de disfunción motora al aparecer lesiones de la motoneurona superior, siendo la espasticidad el principal componente. Las principales manifestaciones son dificultades para caminar, pérdida de destreza, espasmos, hemiparesia. El origen de la espasticidad es generalmente espinal y afecta predominantemente a las extremidades inferiores y al tronco. Con el curso de la enfermedad, los pacientes experimentan un aumento del tono flexor, que puede originar caídas súbitas y espasmos dolorosos en flexión, e imposibilita al paciente hasta el punto de tener que usar una silla de ruedas. Los signos piramidales incluyen la ausencia de los reflejos abdominales, reflejos tendinosos exagerados, reflejos plantares extensores, reflejo mandibular exaltado, y la presencia de reflejos tendinosos clónicos. Los reflejos profundos pueden estar deprimidos o ausentes, especialmente en los brazos. Es también frecuente la fatiga.
Si se ven afectadas las neuronas cerebelosas, se podrán observar por ejemplo una ataxia axial o de tronco (por afectación del vermix cerebeloso), que dará lugar a trastornos del equilibrio y la marcha del paciente, o dismetría en las extremidades (por la afectación del hemisferio cerebeloso ipsilateral a las extremidades afectadas). La disartria no se suele ver al comienzo de la enfermedad.
Visuales (aproximadamente en el 36% de los pacientes)
La neuritis óptica es una frecuente manifestación temprana de la EM, y afecta al menos al 20% de los pacientes. Sus primeros síntomas son normalmente dolor ocular o supraorbitario (a veces acompañado de cefalea) que con frecuencia empeora con el movimiento ocular o se presenta únicamente con dicho movimiento. El dolor se suele acompañar de hipersensibilidad a la presión sobre el globo ocular. La pérdida de visión brusca, generalmente manifestada como visión borrosa, sucede tras el comienzo del dolor. La agudeza visual se recupera generalmente en varias semanas tras el ataque inicial, aunque con peor apreciación del brillo de los colores o intolerancia a la luz brillante. Tras un ejercicio intenso, problemas emocionales, fumar, ingerir una comida caliente, aumento de la temperatura del ambiente, o durante la menstruación, algunos pacientes padecen una recurrencia transitoria de la visión borrosa, una manifestación conocida como fenómeno de Uhthoff. Se cree que del 30% al 80% de los pacientes que sufren un episodio aislado de neuritis óptica y tienen lesiones en la imagen por RM, desarrollarán EM.
Sensitivas (aproximadamente en el 61% de los pacientes)
Los pacientes con EM experimentan una amplia variedad de signos y síntomas sensitivos. Los síntomas puramente sensitivos son frecuentes al comienzo de la enfermedad o en las recaídas iniciales. Son frecuentes las parestesias (hormigueos). En la enfermedad más avanzada, los pacientes pueden sufrir una sensación de constricción en torno a alguna parte de una extremidad inferior o pérdida de la sensibilidad térmica. El síndrome de las piernas inquietas es especialmente frecuente en mujeres.
El signo de Lhermitte es un rasgo clínico frecuente de la EM; se trata de una sensación eléctrica que recorre la espalda en sentido caudal hasta las piernas, cuando el cuello es flexionado. Dicha sensación se da en el 30% o más de los pacientes.
Mentales (aproximadamente en el 40% de los pacientes)
Un deterioro intelectual leve acontece en aproximadamente el 40% de los pacientes con EM, mientras que la discapacidad intelectual grave es mucho menos frecuente. Cuando aparecen los déficits cognitivos, afectan habitualmente al aprendizaje, la memoria reciente, y al proceso de la información más que al lenguaje, que generalmente permanece normal. Los trastornos afectivos son muy frecuentes en pacientes con EM. La depresión es frecuente en la EM; se estima que su incidencia se encuentra entre el 27% al 54%.
2.- ENCEFALITIS DESMIELINIZANTE AGUDA (EDA)
Introducción
Se caracteriza por una respuesta autoinmune, que aparecerá tras una infección; reacción alérgica o post-inmunización.
Se considera que existe una sensibilización de linfocitos contra el tejido encefálico debido a una reacción cruzada entre antígenos infecciosos o agentes de inmunización y antígenos cerebrales, esto produce una repuesta inflamatoria directa que compromete la sustancia blanca del SNC.
Aparece principalmente en adultos jóvenes (20-40 años) y niños (5-15), y raramente aparece antes de los 3 años de edad.
No existe una diferencia de incidencia por sexos. Es más habitual en los países desarrollados.
Clínica
Las infecciones más frecuentes que se asocian a EDA son cuadros virales inespecíficos del tracto respiratorio superior. Para otras infecciones asociadas, ver la tabla 1.
La EDA post-inmunización ha sido descrita después de la inoculación de vacunas contra: sarampión, rabia, rubéola, tifoidea, hepatitis B, difteria, parotiditis y antitoxina tetánica. En el 70% de los casos de los pacientes con EDA diagnosticada, ésta aparece entre 1-4 semanas después de la inmunización.
En la EDA post-infección, los síntomas se inician después de la resolución de un cuadro febril, caracterizándose por; cefaleas, convulsiones y encefalopatía.
La incidencia de la ADE post-sarampión es de 1 paciente por cada 1000 afectados de sarampión (hasta el 20% de éstos, son fatales). La varicela y la rubeola tienen una incidencia de 1 paciente cada 10000-20000 afectados.
Lesiones
Las lesiones en la sustancia blanca son difusas, asimétricas y bilaterales. Se localizan en la región parieto-occipital, corona radiada, cerebelo, tronco del encéfalo, médula espinal y nervios ópticos (ver figura 4).
La sustancia gris, también puede verse comprometida, principalmente a nivel de los ganglios basales y el tálamo.
| Sarampión | Adenovirus |
|---|---|
| Varicela | Hepatitis A y B |
| Rubéola | Citomegalovirus |
| Enterovirus | Chlamidia |
| Epstein-Barr | Legionella |
| HTLV1 | Campilobacter |
| Herpes tipo 6 | Micoplasma pneumoniae |
| Herpes simples | Listeria monocitogenes |
| Influenza A y B | Leptospira interrogans |
Tabla 1. Infecciones asociadas a Encefalitis Desmielinizante Aguda.
Evolución de la EDA
La progresión del cuadro neurológico puede ocurrir en días o semanas, llegando en algunos casos a un mes.
La recuperación presenta tanto un grado como una velocidad variable.
Puede recurrir con infecciones e inmunizaciones, es imprescindible no vacunarse en los 6 meses siguientes a que aparezca.
La mortalidad de los pacientes diagnosticados de EDA, se encuentra entre el 10-20%.
Aproximadamente el 50% de los pacientes diagnosticados de EDA, presentan una completa recuperación. Llegan incluso a recuperarse pacientes que estaban postrados en cama.
Diagnóstico Diferencial
Encefalitis; entre el 90-96% de posibilidades de presentar esta enfermedad.
Mielitis; entre un 5-50% de posibilidades de presentar esta enfermedad.
Polirradiculitis; entre un 1-7% de posibilidades de presentar esta enfermedad.
3.- ESCLEROSIS LATERAL AMIOTRÓFICA (ELA)
Introducción
Trastorno que afecta a la 1ª y 2ª motoneuronas.
Se denomina LATERAL porque están afectadas las segundas motoneuronas situadas en el asta lateral de la médula.
Es un trastorno que aparece fundamentalmente en el adulto, con progresión más o menos lenta pero siempre fatal.
Si afecta a las motoneuronas del bulbo raquideo, aparecerán DISARTRIA y DISFAGIA (líquidos). Se podrá observar la aparición de fasciculaciones y amiotrofía en la lengua.
Etiología
– Aparecen unos 2-3 casos al año por cada 100000 habitantes (en España).
– Enfermedad degenerativa, que puede dividirse en:
— ELA esporádica; sin causas conocidas. Aproximadamente el 90% de los casos.
— ELA familiar; con una perfil autosómico dominante. Aproximadamente el 10% de los casos.
– Afecta principalmente a personas con edades comprendidas entre 45-70 años y con mayor probabilidad a hombres que a mujeres.
– De las enfermedades mencionadas es la que presenta un menor tiempo de evolución.
– La esperanza de vida media de los pacientes es de entre 2 y 5 años desde el inicio de la enfermedad.
Complicaciones en la ELA
– El compromiso de la musculatura respiratoria puede provocar DISNEA.
— Lo que conduce al uso de aparatos para mejorar la ventilación.
– Aparece DISFAGIA para líquidos, lo que conduce a:
— Aumento de la posibilidad de sufrir neumonía por aspiración
— Uso de espesantes en las bebidas.
Diagnóstico de la ELA
– El diagnóstico se realiza mediante la realización de numerosas pruebas que descarten otras enfermedades neurológicas, con la que puede ser confundida, como:
— Lesión medular provocada por un tumor cervical.
— Una artrosis cervical puede dar síntomas parecidos.
— Síntomas comunes con la Esclerosis Múltiple.
– Pruebas para su diagnóstico:
— Resonancia Magnética Nuclear cerebral o espinal.
— Electromiografía (EMG).
— Análisis de orina, sangre y líquido cefalorraquídeo.
Tratamiento de la ELA
– En la actualidad no existe ningún tratamiento viable.
– Se enfoca al tratamiento de los síntomas:
— Espesantes para la DISFAGÍA
— Sonda nasogástrica o gastrostomía
— CPAP u O2, para la dificultad respiratoria.
— Fisioterapia, intentar conseguir la autonomía del paciente a través del ejercicio.
– Tratamiento farmacológico:
— Fármacos antioxidantes; evitan la degeneración neuronal.
— Riluzol: Disminuye la liberación de glutamato. Alarga la vida del paciente unos meses.
3.- LEUCOENCEFALITIS MULTIFOCAL PROGRESIVA (LMP)
Introducción
La leucoencefalopatía multifocal progresiva (LMP) es una enfermedad vírica producida por el papovavirus. Su incidencia en pacientes con SIDA se estima entre el 1- 4%, de manera que hoy día la infección por VIH constituye la patología más frecuentemente asociada a la LMP. Desde el punto de vista histológico, la LMP se caracteriza por la presencia de múltiples áreas de desmielinización, con preservación relativa de axones, y sin infiltrados inflamatorios. Se localizan más frecuentemente en la sustancia blanca de los hemisferios cerebrales, pero pueden afectar al cerebelo, tronco y médula.
Cuadro Clínico
En el inicio está enfermedad es engañosa pudiendo confundirse, por ejemplo con la EM. Son muy frecuentes los defectos de campo visual y la hemiparesia progresiva como primeras manifestaciones.
Además de la hemianopsia unilateral y la hemiparesia que puede progresar hasta la tetraplejía.
La presentación neurológica de la LMP incluye alteraciones en el estado de alerta y capacidades cognitivas, alteraciones en el lenguaje, alteraciones visuales, dificultades con la marcha y el equilibrio, descoordinación de los movimientos de las extremidades.
Diagnóstico
Las lesiones desmielinizantes aparecen con gran intensidad mediante la Resonancia Magnética.
El LCR muestra anomalías inespecíficas. En el EEG suelen aparecer alteraciones discretas e inespecíficas. El diagnóstico definitivo es anatomopatológico, a través de una biopsia cerebral.
Tratamiento
Se han publicado algunos casos de mejoría parcial con citarabina.
4.- SÍNDROME DE GUILLAIN-BARRÉ
Introducción
El síndrome de Guillain-Barré es un trastorno neurológico en el que el sistema inmunológico del cuerpo ataca a una parte del sistema nervioso periférico, aunque también puede afectar al sistema nerviosos central.
Puede aparecer de forma muy brusca e inesperada. El trastorno puede desarrollarse a lo largo de unos días, o puede tardar hasta varias semanas. El paciente está más débil en las dos primeras semanas después de la aparición de los síntomas.
Se asocia con infecciones virales, gastrointestinales y/o del tracto respiratorio superior. También existen evidencias de un mecanismo autoinmune, que aparece después de una infección por campylobacter jejuni.
Esta enfermedad aparece en mayor proporción en adultos jóvenes (de 20-40 años) y existe influencia por sexos, siendo más afectados por esta enfermedad los varones que las mujeres.
Es una enfermedad poco común, ya que afecta a 1 persona por cada 100.000 habitantes.
Clínica
Aparecen tetraparesia flácida y arreflexia.
El paciente comienza a sentir debilidad, comienza en los miembros inferiores y asciende de forma progresiva hasta que afecta a todo el cuerpo. Estos síntomas pueden aumentar en intensidad, de modo que los músculos no puedan utilizarse y el paciente queda casi totalmente paralizado. Cuando esto ocurre, la vida del paciente se encuentra en peligro ya que existe una posibilidad real de que se interfiera con la respiración y/o el ritmo cardíaco.
Afecta a ambos lados, manteniendo una simetría en cuanto a las estructuras afectadas.
Es muy extraño que aparezca atrofia muscular en los pacientes con esta enfermedad.
Evolución
Existe una rápida progresión de la debilidad muscular, siendo habitual que ocurra en el plazo de 4 semanas.
Los pacientes presentan recuperación entre la 2ª-4ª semana desde que terminase la progresión de la debilidad muscular, la recuperación observada puede durar meses.
El 50% de los pacientes diagnosticados del síndrome de Guillain-Barré, permanece con alguna secuela. Mientras que el 5% de los pacientes diagnosticados del síndrome de Guillain-Barré, mueren.
5.- POLINEUROPATIA DESMIELINIZANTE CRÓNICA
Introducción
Se define como una afección neuropática simétrica de predominio motor con debilidad proximal y distal, asociada a pérdida de los reflejos osteotendinosos de curso progresivo o recurrente. Cursa con aumento del contenido de proteínas en el líquido cefalorraquídeo, así como evidencias de desmielinización.
Esta enfermedad aparece a una edad avanzada, siendo frecuente encontrarla en paciente con 50-60 años de edad.
Presenta un curso crónico, que en ocasiones presenta recurrencias de forma intermitente.
Tiene una mayor afectación sensitiva.

La ataxia,es una enfermedad polineuropatía desmielizante, puede recuperar la mielina,?