Curso de farmacogenética y su aplicación clínica en la epilepsia farmacorresistente
Manual de formación Continuada 2018-2019. Hospital de la Princesa
Desirée Nava Cedeño; Concepción Alonso Cerezo; Paloma Pulido Rivas; Rafael García Sola
Hospital Universitario de La Princesa. Madrid.
1. INTRODUCCION
El presente tema tiene como objetivo realizar una revisión actual de los avances de la farmacogenética en la epilepsia farmacorresistente (EF), haciendo referencia a los aspectos epidemiológicos, definiciones actualizadas, bases moleculares e hipótesis de los polimorfismos genéticos involucrados en los mecanismos fisiopatológicos de resistencia. Se excluyen de esta revisión el análisis de las variables genéticas causales y de las relacionadas con reacciones adversas provocadas por los fármacos antiepilépticos (FAE).
Dado que la epilepsia constituye una patología de alta prevalencia e incidencia, los FAE presentan una gran variabilidad interindividual en su respuesta y un 25% de los pacientes son refractarios al tratamiento (1), por lo que la farmacogenética se convierte en una herramienta fundamental para el abordaje de esta patología.
La epilepsia es uno de los trastornos neurológicos crónicos más prevalentes mundialmente que afecta a personas de todos los grupos étnicos, en todas las áreas geográficas y de todas las clases sociales. Según la Organización Mundial de la Salud (OMS), 50 millones de personas padecen o han padecido de epilepsia (2). Se calcula que la población con epilepsia activa, aquella que presenta alguna crisis en los últimos cinco años de vida (3), está en torno a 8 por cada 1.000 habitantes. Bell y colaboradores en 2014, realizan un análisis sobre la prevalencia de epilepsia activa en Europa encontrando cifras de 6 por cada 1.000 habitantes (4). Según informe de la Fundación Española de Enfermedades Neurológicas (FEEN), en 2010 en España había 360.000 casos de epilepsia y la incidencia anual era de 34 casos por cada 100.000 habitantes, es decir, 17.000 casos nuevos cada año, por lo que la epilepsia es la enfermedad neurológica más prevalente del país y por lo tanto constituye un problema de salud pública (5). En la actualidad no existen datos precisos de EF en España por la falta de estudios epidemiológicos recientes. Sin embargo, un estudio publicado en la Revista de Neurología en 2008 puede ser orientativo. Este se realizó en consultas ambulatorias de neurología y especializadas de epilepsia, detectando una prevalencia de EF del 22,7% (6).
La mayoría de los pacientes epilépticos se pueden controlar con FAE, pero entre un 20 a 30% no responden al tratamiento convencional (1), por lo que son candidatos a tratamientos alternativos como la neuroestimulación del vago o cirugías como la amigdalo-hipocampectomía. Estos porcentajes se han mantenido a lo largo de los años, a pesar de contar con nuevos FAE con mejores perfiles farmacocinéticos y algunos de ellos con nuevos mecanismos de acción (7). A la observación de estos resultados parece que la farmacorresistencia tiene un sustrato fisiopatológico que no depende de la estructura química, ni de propiedades farmacocinéticas o farmacodinámicas del fármaco sino de un eslabón común a todos ellos. En consecuencia, probablemente el mecanismo de farmacorresistencia no es selectivo a un fármaco, por lo que deberíamos plantearnos hipótesis que no impliquen farmacoselectividad.
2. DEFINICIÓN DE EPILEPSIA FARMACORRESISTENTE
La Liga Internacional contra la epilepsia (International League Against Epilepsy, ILAE) en 2010 propone por primera vez una definición consensuada de EF la cual consiste en aquella epilepsia que diagnosticada correctamente presenta un mal control de las crisis, a pesar de la utilización de dos esquemas de FAE apropiados para el tipo de epilepsia, en monoterapia o terapia combinada y en dosis máximas tolerables, durante un tiempo suficiente para asegurar su ineficacia. Además, el paciente debe haber realizado el tratamiento indicado (8).
- Número de esquemas terapéuticos: está implícito que los fármacos que se utilicen hayan demostrado su eficacia en ensayos clínicos adecuadamente diseñados con el más alto grado nivel de evidencia (aleatorizados y controlados) para el tipo de epilepsia que padezca el paciente (9). La decisión de dos esquemas terapéuticos se basa en estudios que demostraron la baja probabilidad de que al asociar otro nuevo fármaco logre que el paciente alcance la libertad de crisis epilépticas. El más reciente de ellos, realizado por Brodie y colaboradores en 2012 arrojaba los siguientes resultados: el 49,5% de los pacientes está libre de crisis después del primer régimen de FAE y solo el 13,3% y el 3,7% logran el control de las crisis después del segundo y tercer régimen, respectivamente (10). Por lo que la respuesta al primer y segundo FAE es altamente predictiva del desenlace terapéutico.
- Tiempo: con respecto al tiempo suficiente como para asegurar la ineficacia de un fármaco antiepiléptico, es uno de los tópicos que más controversia ha generado en el entorno médico. La ILAE llega a estandarizar este tiempo estableciendo como epilepsia controlada o libertad de crisis aquella condición en la que hay ausencia de crisis epilépticas durante al menos tres veces el período máximo que el paciente ha estado sin crisis durante el último año o la ausencia de crisis durante 12 meses; cualquiera de los dos criterios que constituya el período de tiempo más largo. Por ejemplo, si un paciente presenta un periodo máximo libre de crisis de tres meses, debería estar sin crisis durante 9 meses después de la nueva intervención para considerar que se encuentra con una epilepsia controlada. Pero si el paciente ha estado 12 meses sin crisis epilépticas, se considera el segundo criterio por ser el período más largo.
- La ILAE hace énfasis en no catalogar de EF a aquella en la que el paciente suspenda el tratamiento por sus efectos secundarios y en la que no hubiese adherencia al tratamiento (8).
- Por otra parte, cuando dos medicamentos que se administran simultáneamente se metabolizan por el mismo citocromo y uno de ellos es un inductor enzimático, este se unirá al sitio activo de la enzima aumentando su actividad por lo que provocará una disminución de la biodisponibilidad del otro fármaco debido al aceleramiento del metabolismo enzimático. Este fenómeno no se puede catalogar de farmacorresistencia, sino de interacción farmacológica y debe ser tomado en cuenta en la práctica clínica, de ahí que se le dé prioridad a utilizar los nuevos antiepilépticos con mejor perfil farmacocinético y menos interacciones farmacológicas.
3. HIPÓTESIS SOBRE LOS MECANISMOS FISIOPATOLÓGICOS DE LA EPILEPSIA FARMACORRESISTENTE
Actualmente no se conocen todos los mecanismos fisiopatológicos por los que dos pacientes con una misma etiología, con crisis epilépticas similares y tratados con los mismos fármacos a dosis iguales responden de manera diferente. Así mismo, no está del todo claro porque unos pacientes inicialmente son farmacosensibles y posteriormente se hacen farmacorresistentes. Esta variabilidad interindividual y en un mismo individuo en la respuesta al tratamiento farmacológico podría ser explicada en parte por la presencia de polimorfismos genéticos. Un polimorfismo genético es una variable genética que aparece con una frecuencia igual o mayor del 1% en la población general (11). En este sentido, la farmacogenética es la disciplina que trata de determinar las variantes genéticas entre los pacientes farmacorresistentes y farmacosensibles para identificar su efecto sobre la respuesta a los FAE con el objetivo de predecir la farmacorresistencia y realizar una prescripción personalizada, o en otras palabras, que el Clínico recete un antiepiléptico adecuado y a dosis ajustadas en relación a la genética del paciente que se esté tratando.
Cualquier clasificación sería arbitraria, pero, para facilitar el estudio de esta condición, se proponen las siguientes hipótesis para explicar el mecanismo fisiopatológico de la farmacorresistencia según el esquema representado en la Figura 1.
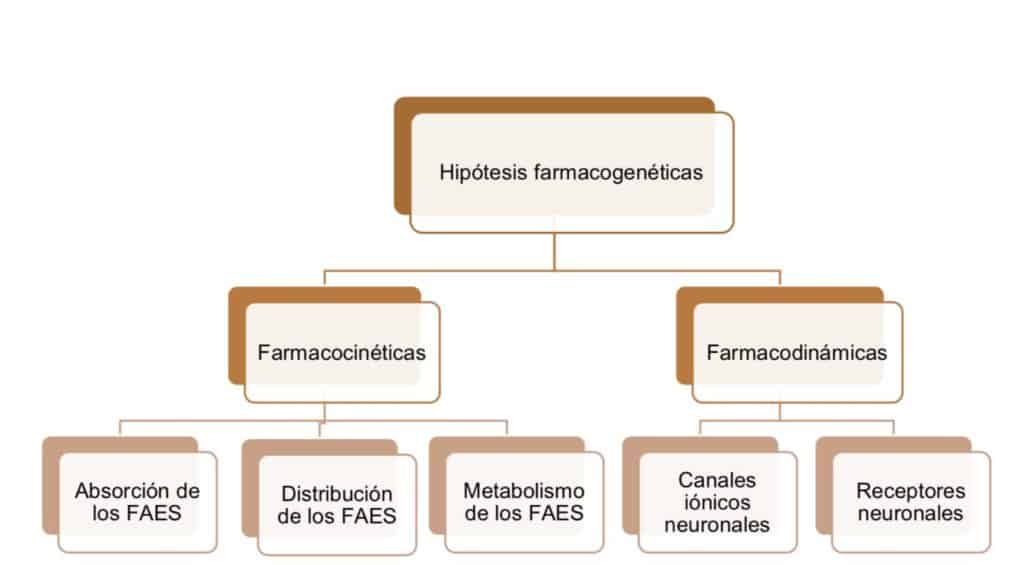
Este esquema no implica necesariamente que las hipótesis planteadas recojan todos los posibles mecanismos de farmacorresistencia, ni tampoco que estén identificados polimorfismos relevantes en todas las hipótesis formuladas; es más bien una manera de aproximarse a todas las posibilidades que podrían explicar la farmacorresistencia en los pacientes epilépticos.
Hipótesis farmacocinéticas: la farmacocinética es una rama de la farmacología que estudia el paso de los fármacos a través del organismo en función del tiempo y la dosis. Comprende los procesos de absorción, distribución, metabolismo y eliminación (12).
- Hipótesis farmacocinética de la absorción de los FAE: afecta a los polimorfismos en los genes que codifican las enzimas metabolizadoras de drogas en el tracto gastrointestinal, lo cual podría disminuir la biodisponibilidad del fármaco (13).
- Hipótesis farmacocinética de la distribución de los FAE: comprende los polimorfismos en los genes que codifican las proteínas transportadoras de fármacos en la barrera hematoencefálica (BHE), lo cual repercutiría en la biodisponibilidad del ́rmaco antiepiléptico en el foco epileptógeno (14-17).
- Hipótesis farmacocinética del metabolismo de los FAE: incluye los polimorfismos genéticos que provocan ganancia de función en las proteínas metabolizadoras, ocasionando que el paciente portador no alcance los niveles séricos terapéuticos (18).
Hipótesis farmacodinámicas: la farmacodinamia se define como la rama de la farmacología que estudia los mecanismos de acción de los fármacos y sus efectos bioquímicos sobre el organismo (12), a través de su acción sobre moléculas diana. En el caso de los FAE, los principales blancos son los canales iónicos neuronales de sodio, calcio y potasio así como también receptores neuronales de los neurotransmisores más importantes involucrados en la epilepsia: ácido gamma-aminobutírico (GABA) y glutamato.
3.1 Hipótesis farmacocinéticas
3.1.1 Hipótesis farmacocinética de la absorción de los FAE: polimorfismos genéticos en enzimas metabolizadoras de fármacos en los enterocitos
Una de las propiedades físico-químicas de la mayoría de los FAE es que son lipofílicos y por lo tanto, penetran las membranas por transporte pasivo. Sin embargo, su absorción a través del enterocito en la mucosa gastrointestinal podría estar limitada por polimorfismos en los genes que codifican enzimas metabolizadoras como CYP3A4 y CYP2C9 y por ende, disminuir la biodisponibilidad del fármaco. Hasta el momento no se han publicado estudios que hayan identificado un efecto clínicamente significativo (13).
3.1.2 Hipótesis farmacocinética de distribución de los FAE: polimorfismos genéticos en proteínas transportadoras de fármacos en el parénquima cerebral
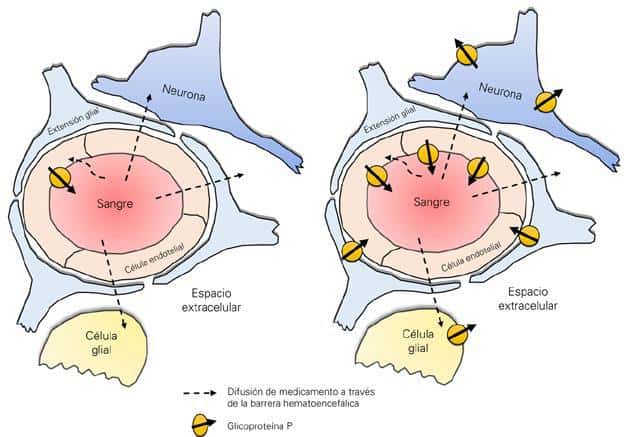
En el sistema nervioso central (SNC) existen proteínas transportadoras de fármacos que se encuentran en la membrana luminal del endotelio vascular de la BHE, las cuales se encargan de la protección de las neuronas por medio de la expulsión al torrente sanguíneo de sustancias tóxicas, incluidas entre ellas a muchos fármacos. La glicoproteína P (gp-P), también denominada MDR1, es una bomba transportadora de fármacos que se expresa normalmente en la BHE. La sobreexpresión adquirida o intrínseca de esta proteína hace que esta se exprese en astrocitos y neuronas, ocasionando la expulsión del FAE y disminuyendo así la concentración terapéutica del mismo en el foco epileptógeno (14). En consecuencia, generaría resistencia a todos aquellos fármacos que sean sustrato de la gp-P y explicaría porque algunos pacientes, aun teniendo niveles séricos terapéuticos, presentan farmacorresistencia. Incluso, explicaría aquellos pacientes que no responden a FAE que son estructuralmente diferentes y presentan distintos mecanismos de acción.
En el cromosoma 7q21 se encuentra el gen ABCB1 (o MDR1), el cual codifica para la gp-P. Este gen se compone de 28 exones y se han descrito múltiples polimorfismos genéticos que podrían estar involucrados en el mecanismo de farmacorresistencia. De todos ellos, el más estudiado es el polimorfismo de un solo nucleótido (rs1045642) que se encuentra en el exón 27 (NM_000927), en el cual hay una sustitución de una timina por una citosina en la posición 3435. Se trata de un SNP de tipo sinónimo o silencioso ya que no modifica la secuencia de aminoácidos de la proteína resultante (16). Aunque a priori podemos pensar que los SNP involucrados en el desarrollo de la farmacorresistencia deberían ser de alto impacto, es decir, que ocasionarían alteración en la estructura de la proteína por un cambio de aminoácido o en el marco de lectura o que la truncarían por un codón de terminación prematuro, este tipo de SNP están mayormente relacionados con ser causantes de enfermedad. Por el contrario, los SNP de tipo silencioso parecen jugar un papel importante en la farmacogenética ya que pueden afectar al proceso de transcripción, ocasionar inestabilidad en el ARN mensajero, o bien estar en desequilibrio de ligamiento con otros SNP no silenciosos que pudiesen explicar el fenotipo, y por la evidencia científica parecen estar más relacionados con la variabilidad en la respuesta farmacológica.
Esta es una de las hipótesis más estudiadas pero también una de las que ha arrojado más resultados controvertidos y con poca evidencia en la actualidad. Desde el año 2003 en que se describió la asociación de este polimorfismo con EF, se han publicado numerosos estudios que intentan comprobar este supuesto, sin embargo, los resultados han sido contradictorios y no reproducibles. Algunos estudios concluyen que el genotipo asociado a EF es el TT, otros el CC, mientras que la mayoría no encuentran ninguna asociación. No obstante, un metanálisis publicado en 2015 que incluyó 8.604 sujetos de 30 estudios encontró significación estadística entre el polimorfismo rs1045642 y la EF (16), por lo que aún no se puede descartar esta teoría. Es necesario realizar estudios metodológicamente mejor diseñados con tamaños muestrales más grandes que incluyan definiciones claras y consensuadas de EF y grupos de estudio más homogéneos en cuanto a diagnóstico, fármacos utilizados y origen étnico, para poder concluir definitivamente la contribución genética de este polimorfismo en la EF. Por otro lado, existen estudios que analizan haplotipos conformados por varios SNP, en lugar de analizar un único polimorfismo. Estos trabajos analizaron el ya nombrado rs1045642 y otros dos SNP que aparecen con mayor frecuencia en la población, el rs1128503 (p.Gly412Gly) y el rs2032582 (p.Ala893Ser/Thr), pero sin llegar a ser concluyentes (17).
Se han observado asociaciones entre otros genes que codifican proteínas transportadoras y EF, como el ABCC2, pero con menor significancia estadística que con ABCB1 (17).
En la Figura 2 se puede visualizar una representación esquemática del planteamiento que propone la hipótesis farmacocinética a nivel de los transportadores de FAE en las neuronas y astrocitos para explicar la farmacorresistencia.
3.1.3 Hipótesis farmacocinética del metabolismo de los FAE: polimorfismos genéticos en proteínas metabolizadoras de fármacos
El metabolismo de los FAE está mediado principalmente por el citocromo P450 (CYP P450). Este constituye una superfamilia enzimática involucrada en el metabolismo oxidativo de los compuestos endógenos como son los ácidos grasos, el colesterol, las vitaminas liposolubles y las hormonas esteroides. Además, participa en el metabolismo de sustratos de naturaleza exógena y lipofílica como fármacos, procarcinógenos y anestésicos, entre otros. Este complejo enzimático se encuentra presente principalmente en las mitocondrias y retículo endoplásmico liso de tejidos como riñón, pulmón, piel, intestino, cerebro, corteza adrenal,testículos y placenta, pero es en el hígado donde tiene su mayor expresión. Desde el punto de vista bioquímico es un sistema de monooxigenasas dependiente de NADPH en un complejo multienzimático cuya oxidasa final es una hemoproteína denominada CYP P450. La denominación proviene de que esta hemoproteína en su forma reducida presenta un máximo de absorbancia a los 450 nm. Este sistema enzimático cataliza reacciones en donde solo se incorpora uno de los átomos de oxígeno molecular y el otro se reduce a agua. Esto tiene como consecuencia que se produzca hidroxilación (reacciones de fase I) del sustrato, lo que ocasiona la formación de productos más polares, solubles en agua y derivados farmacológicos menos potentes que pueden ser excretados directamente o en algunos casos tras ser conjugados con otros compuestos metabólicos (reacciones de fase II). Debido a la complejidad de esta familia de enzimas, se denominan con las siglas CYP seguido de un número que corresponde a la familia, una letra que identifica a la subfamilia y otro número que corresponde al gen.
Existen 4 grandes familias del CYP P450 que están involucradas en el metabolismo de los FAE. Variantes alélicas que codifican isoenzimas con actividad diferente, ya sea a través de la inducción o inhibición de los sistemas enzimáticos microsomales hepáticos, podrían afectar a las concentraciones séricas de los FAE y por ende, a su eficacia y tolerabilidad clínica. Polimorfismos genéticos que codifiquen isoenzimas con ganancia de función ocasionará que el individuo portador metabolice más rápidamente el fármaco y por lo tanto no alcance niveles séricos terapéuticos requiriendo dosis mayores del fármaco para lograr eficacia clínica. Por otro lado, polimorfismos genéticos que codifiquen isoenzimas con disminución de la función se traducirán en que el individuo portador metaboliza más lentamente con el subsecuente riesgo de presentar niveles tóxicos de los FAE.
La mayoría de los FAEs son metabolizados por el CYP3A4 el cual muestra 30 polimorfismos, de los cuales los más caracterizados son CYP3A4*1B (rs2740574) y CYP3A4*22 (rs2687116). Un reciente estudio incluyendo 23 pacientes resistentes y 7 respondedores demostró que la variante alélica CYP3A4*1B es un factor de riesgo para desarrollar resistencia en población pediátrica (18). Se deben validar estos resultados con otros estudios de investigación dado el escaso número de pacientes incluidos.
Otra familia importante involucrada en el metabolismo de los FAE, es la familia del CYP2. Los polimorfismos en CYP2C9, CYP2C19 y CYP2D6 han sido estudiados ampliamente en una gran cantidad de poblaciones pero sin llegar a resultados estadísticamente significativos. Las principales variantes del alelo silvestre CYP2C9*1 (rs1057910) son CYP2C9*2 (rs1799853) y CYP2C9*3 (rs1057910), los cuales están asociados a una actividad reducida enzimática con respecto a la variable silvestre, por lo que está muy bien definido su papel en las reacciones adversas pero en cuanto a resistencia no hay resultados claros. Respecto al CYP2C19 se han descrito 28 variantes, pero los más ampliamente descritos son el CYP2C19*2 (rs4244285), el cual causa una deleción de 40 nucleótidos con el subsecuente cambio de marco de lectura, y CYP2C19*3 (rs4986893), el cual conduce a un codón de parada prematura dando como resultado la producción de una proteína truncada, por lo que estas dos isoenzimas producen disminución o ninguna actividad enzimática. En el estudio anteriormente citado (18), la variante alélica heterocigótica del rs4244285 se presentó con mayor frecuencia en los pacientes epilépticos resistentes a FAE que la o la silvestre.
La variante alélica rs2606345 en CYP1A1 en mujeres con epilepsia en una población india se asoció con una respuesta deficiente a los FAE, aunque estos resultados se deben validar en otras poblaciones.
Un factor a considerar es la frecuencia alélica de estos polimorfismos en las poblaciones estudiadas, puesto que los resultados de una población africana no se pueden extrapolar a una población europea debido a las diferencias en la variabilidad genética, de ahí la importancia de realizar estudios en nuestra población.
Hasta el presente no existe evidencia de que la genotipificación de los citocromos mejore el resultado del tratamiento con FAE en comparación con la observación clínica y la determinación de la concentración sérica del fármaco.
Cabe destacar que esta hipótesis no explica los pacientes farmacorresistentes con niveles séricos adecuados y tampoco contempla un sustrato fisiopatológico de un citocromo común en el metabolismo de todos los FAE que explique la farmacorresistencia no selectiva.
3.2 Hipótesis farmacodinámicas: polimorfismos en los genes que codifican las moléculas diana de los FAE
Para comprender el planteamiento de estas hipótesis, debemos distinguir entre variantes genéticas causales y polimorfismos genéticos que expliquen la variabilidad en la respuesta al tratamiento. Generalmente, las variantes genéticas patogénicas se presentan con una frecuencia muy baja en la población general (<1.000.000), se encuentran en residuos altamente conservados del genoma humano y en los algoritmos de predicción in silico suelen encontrarse en dominios de la proteína que alteran su función. En cambio, los polimorfismos genéticos, por definición, se presentan en la población con una frecuencia mayor al 1% y los relacionados con farmacogenética mayoritariamente son SNP que pueden estar en regiones exónicas o intrónicas, alterar la cadena de aminoácidos, es decir, no sinónimos (missense), o ser sinónimos (synonymous). Existen guías consensuadas para interpretar y clasificar las variantes causales como, por ejemplo, la publicada en 2015 por el Colegio Americano de Genética Médica y Genómica (ACMG) (19), sin embargo, aún no existen guías estandarizadas para la clasificación de las variables farmacogenéticas.
Es importante diferenciar si la farmacorresistencia observada se debe a una variante causal, como por ejemplo sucede en el síndrome de Dravet por alternaciones en el gen SCN1A (codifica la subunidad alfa-1 del canal de sodio neuronal), en la encefalopatía de inicio temprano debido a una ganancia de función en el gen KCNT1 (codifica el canal voltaje dependiente de potasio en el SNC) y en el amplio espectro de epilepsias causadas por alteraciones en el gen GRIN2A (codifica una subunidad del receptor N-metil-D-aspartato, NMDA). Estos síndromes tienen en común que son causados por variantes genéticas patogénicas en dianas terapéuticas de FAE y que cursan con farmacorresistencia. Sin embargo, en estos pacientes la resistencia se explica por la etiopatogenia de la epilepsia y no porque sean portadores de polimorfismos genéticos asociados a su tipo de epilepsia que les confieran resistencia. Cabe destacar que este tipo de epilepsias de etiología genética podrían ayudar a dilucidar el posible mecanismo de farmacorresistencia en aquellas epilepsias de origen no genético.
Por otra parte, es importante recordar la fisiopatología de la epilepsia para comprender cómo variantes alélicas en los canales iónicos y receptores de los neurotransmisores pueden estar involucrados en mecanismos de farmacorresistencia (Figura 3).
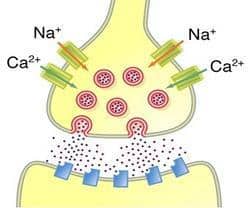
Los impulsos nerviosos son en realidad impulsos eléctricos llamados potenciales de acción, los cuales se desplazan lo largo de la red neuronal debido al flujo de iones que entran y salen a través de los canales iónicos proteicos transmembrana. Estos impulsos están controlados por los neurotransmisores en el axón terminal de la neurona presináptica. Los neurotransmisores son liberados al espacio sináptico y actuarán en los receptores de la neurona post sináptica. Si la neurona presináptica es gabaérgica, el impulso nervioso será de tipo inhibitorio, es decir, se liberará GABA al espacio sináptico que interactuará con el receptor postsináptico GABA-A. Este receptor es un canal iónico activado por ligando, por lo que la unión de las moléculas de GABA a sus sitios de unión en la parte extracelular del receptor desencadenará la apertura de un poro selectivo de iones de cloruro. El aumento de la conductancia del cloruro repolariza la membrana neuronal por lo que inhibe el disparo de nuevos potenciales de acción . En caso de ser un impulso excitatorio la neurona presináptica será de tipo glutaminérgica y la entrada de calcio intracelular mediará la liberación de las vesículas cargadas de glutamato, el cual al ser liberado a la hendidura sináptica interactuará con los receptores neuronales excitatorios principales como NMDA o el ácido α-amino-3- hidroxi-5-metilo-4-isoxazolpropiónico (AMPA), los cuales son canales iónicos que mediarán la entrada de cationes como el calcio y el sodio, respectivamente, los cuales producirán en la neurona postsináptica la despolarización de la membrana conduciendo el impulso eléctrico.
El desequilibrio entre los impulsos excitatorios e inhibitorios a favor de una mayor excitabilidad en la corteza cerebral es la base fisiopatológica por lo cual se produce la epilepsia. Las variantes alélicas en los genes que codifican los receptores de sodio de las neuronas presinápticas gabaérgicas y que se traduzcan en una pérdida de función, producirán un aumento de la excitación a nivel del SNC y se manifestarán clínicamente en forma de convulsiones intratables con fármacos que tengan como diana terapéutica estos canales. La ganancia de función de los canales de sodio en neuronas glutaminérgicas traducirá un desequilibrio de los impulsos nerviosos a favor de una mayor excitabilidad y ocasionarán que la mayoría de los fármacos disponibles en la actualidad no puedan disminuir eficazmente el inicio de las convulsiones. Estos son planteamientos hipotéticos, ya que hasta la actualidad, estos mecanismos no están demostrados con significancia estadística y mucho menos clínica y, como veremos más adelante, no explican toda la heterogeneidad de resistencias que se observan en los pacientes.
Las hipótesis farmacodinámicas tienen importantes críticas, entre ellas: no se conocen con exactitud todos los mecanismos de acción de los FAE, un mismo FAE puede tener varias dianas terapéuticas y un paciente puede recibir tratamiento con fármacos con diferentes mecanismos de acción.
3.2.1 Hipótesis farmacodinámica para polimorfismos genéticos en los canales iónicos neuronales presinápticos
Los canales de sodio regulados por voltaje son uno de los objetivos principales de varios FAE, por lo que mucha de la investigación farmacogenética se ha dirigido a analizar genes como el SCN1A, SCN2A y SCN3A que codifican subunidades alfa del canal de sodio altamente expresado en SNC. Sin embargo, los hallazgos hasta la fecha han sido inconsistentes (17). Los bloqueadores de canales de sodio han demostrado una eficacia significativa en las encefalopatías epilépticas ocasionadas por variantes patogénicas en estos genes. El mecanismo fisiopatológico del efecto aún no se ha dilucidado pero parece estar presente fundamentalmente en pacientes con variaciones con ganancia de función (16).
3.2.2 Hipótesis farmacodinámica para polimorfismos genéticos en los receptores postsinápticos neuronales
El gen GABRA1 codifica la subunidad alfa-1 del receptor de GABA. Este es un canal de cloruro activado por ligando. Las alteraciones en este gen están relacionadas con epilepsia temporal resistente (1).
3.3 Otras hipótesis no farmacogenéticas
- Hipótesis inmunológica: debido a la identificación de inmunoglobulinas contra receptores neuronales excitatorios NMDA e inhibitorios GABA tipo A (GABA-A) en pacientes que padecieron infecciones en el SNC asociadas a convulsiones, las cuales posteriormente progresaron a epilepsias resistentes, se ha planteado la hipótesis de que la farmacorresistencia se puede deber a la formación de anticuerpos (1).
- Hipótesis sobre el mecanismo de acción de los FAE: esta hipótesis propone que los FAE no actúan sobre los mecanismos fisiopatológicos involucrados en la generación de las crisis epilépticas como el estrés oxidativo, la disfunción mitocondrial o la respuesta hiperinmune con disfunción de la BHE, sino que simplemente inhiben el exceso de impulsos nerviosos excitatorios mediados por el glutamato y sus receptores para el NMDA y AMPA y potencian los impulsos nerviosos inhibitorios mediados por el GABA y su receptor principal, previniendo de esta forma la aparición de las crisis epilépticas, pero no actuando sobre lo que las causa (18).
- Hipótesis fisiopatogénicas: sugiere que las variantes genéticas causales en los genes que codifican las proteínas involucradas en la patogenia del tipo de epilepsia que padece el paciente explican la farmacorresistencia (19).
- Hipótesis de la severidad intrínseca: postula que la farmacorresistencia resulta de la disfunción aumentada de las vías biológicas que subyacen a la epilepsia (20).
4. LIMITACIONES ACTUALES DE LA FARMACOGENÉTICA EN LA PRÁCTICA CLÍNICA
- El desconocimiento de todos los genes involucrados en las epilepsias: los genes causales se han logrado identificar principalmente en las epilepsias monogénicas, que representan sólo el 1 a 2% de los síndromes epilépticos. Esperemos que en un futuro próximo se pueda disponer de una clasificación de las epilepsias que incluya además de la semiología de las crisis y el electroencefalograma típico, también las variantes genéticas causantes de la enfermedad.
- La presencia de epilepsias poligénicas: hace más difícil determinar el mecanismo por el cual sucede la enfermedad.
- La no correlación directa entre genotipo y fenotipo: una misma variante genética patogénica puede producir un amplio abanico de manifestaciones clínicas o un fenotipo ser producido por variaciones genéticas en distintos genes. Por ejemplo, las variantes patogénicas en el gen SCN1A pueden ocasionar desde convulsiones febriles plus (GEFS+) hasta la epilepsia mioclónica severa de la infancia o síndrome de Dravet. Las crisis simples en pacientes pediátricos en el contexto de fiebre se consideran normales si se presentan desde los 6 meses hasta los 6 años, tienen una duración corta y normalmente no cursan con estatus epilépticos. Las convulsiones febriles plus (GEFS+) se caracterizan por extenderse más allá de los 6 años prolongarse en la duración más que una convulsión febril simple hasta en algunos casos evolucionar a un estatus epiléptico y/o desencadenarse sin que el paciente esté febril o con ligeras variaciones de temperaturas con un curso benigno. La epilepsia mioclónica severa de la infancia cursa con crisis epilépticas de diversas semiologías: tónico-clónico generalizadas, parciales secundariamente generalizadas, ausencias, y frecuentemente llegan a estatus epilépticos resistentes al tratamiento farmacológico y asociadas a deterioro psicomotor irreversible con un curso progresivo.
- La complejidad del genoma humano: las variables genéticas no actúan aisladamente, sino en el contexto del resto del genoma, interaccionan con otros genes y los genes con el ambiente. Así, hay que tener en cuenta fenómenos de epistasis resultantes de la interacción entre diferentes genes al expresar un determinado carácter fenotípico. Por lo tanto, con el fin de diseñar fármacos dirigidos, es importante no solo identificar la variante genética causal involucrada en la característica fenotípica de farmacorresistencia si no determinar la alteración funcional subyacente producida por esta en la red neuronal, resultante de la suma de los efectos independientes de cada variable genética con la variante causal en el individuo.
- El impacto en la estabilidad de la red neuronal que puede significar una variable genética: cada individuo según su perfil genético tendrá una variabilidad individual y esto determina su red de excitabilidad específica, alterando toda la red neuronal y no sólo un área del cerebro.
- La heterogeneidad en la epilepsia y patrones clínicos de resistencia: la epilepsia se compone de un conjunto heterogéneo de enfermedades con un sustrato fisiopatológico común pero con diversas etiologías y manifestaciones clínicas. En los pacientes epilépticos se pueden observar distintos patrones de resistencia. Algunos son resistentes de novo, es decir, desde el inicio las crisis epilépticas son resistentes al tratamiento farmacológico. Otros presentan resistencia progresiva, inicialmente tienen crisis epilépticas que responden al tratamiento farmacológico durante varios años y posteriormente evolucionan a crisis que no responden a los FAE, sin volver a presentar nuevamente remisión. Otro de tipo de patrón de presentación es el de recaída y remisión, que consiste en que la respuesta terapéutica de los pacientes fluctúa entre periodos de farmacorresistencia y farmacosensibilidad. Así, sería desacertado pensar que todos estos patrones son explicados por los mismos mecanismos de farmacorresistencia.
- Falta de conocimiento de todos los mecanismos de acción y dianas terapéuticas de los antiepilépticos.
- Comprender todos los mecanismos fisiopatológicos involucrados en la epilepsia farmacorresistente y la historia natural de esta.
- Carencia de formación en la interpretación de variables genéticas y su aplicación en la clínica.
- Ausencia de guías y directrices estandarizadas para la interpretación de variables farmacogenéticas.
5. CONCLUSIONES
Por todo lo expuesto anteriormente, podemos concluir que la epilepsia farmacorresistente es una condición altamente prevalente y la respuesta a los FAE tiene un importante grado de variabilidad intra- e interindividual. Esta gran variabilidad puede ser el fundamento biológico de la farmacorresistencia. Es difícil explicar la farmacorresistencia en epilepsia con un único modelo fisiopatológico, por lo que se debe pensar en esta condición como el resultado de múltiples interacciones neurobiológicas entre variables relacionadas con la patología de base, con el fármaco antiepiléptico y con los aspectos farmacogenéticos propios de cada individuo. Desde el punto de vista molecular, se plantean algunas hipótesis que intentan explicar el fenómeno de farmacorresistencia en epilepsia. Dentro de estas hipótesis, la más reconocida actualmente es la hipótesis de los transportadores, puesto que explicaría los pacientes que son resistentes a diversos medicamentos independientemente de sus mecanismos de acción, propiedades farmacocinéticas y farmacodinámicas, además de responder a la interrogante de los pacientes con niveles séricos adecuados que no tienen una eficaz respuesta a los FAE. Sin embargo, una importante crítica a esta hipótesis es que no todos los FAE son sustrato de la glicoproteína P. Por tanto, aún es necesario continuar realizando investigación con ensayos multicéntricos, con un número adecuado de pacientes, estratificados según la etiología, el tipo de epilepsia, el patrón de resistencia y los fármacos utilizados, y en donde se incluyan todas las posibles hipótesis que pudiesen explicar esta condición.
En la actualidad, ninguno de los mecanismos fisiopatológicos planteados responde a todas las interrogantes observadas en los pacientes epilépticos farmacorresistentes y tampoco ninguno de los polimorfismos genéticos estudiados asociados a la farmacorresistencia en la epilepsia ha alcanzado evidencia significativa para trasladarse a la práctica clínica.
Entender los mecanismos fisiopatológicos implicados en la farmacorresistencia e identificar las diferencias genéticas existentes entre los pacientes farmacosensibles y farmacorresistentes permitirá:
- Determinar un biomarcador genético que permita predecir la farmacorresistencia y por lo tanto, realizar un diagnóstico precoz de EF, aumentando la calidad de vida y salud de los pacientes y reduciendo así los costes en la sanidad asociados a un uso inadecuado de los fármacos.
- Desarrollar nuevos tratamientos dirigidos a otras dianas terapéuticas o mejorar los ya existentes.
- Indicar el tratamiento según los SNP presentes en el paciente para tratar a los enfermos y no las enfermedades, evolucionando hacia una medicina personalizada.
A pesar de las importantes implicaciones que podría tener en la práctica clínica, todavía queda un camino largo por recorrer en estas áreas del conocimiento. Es, por tanto, necesario desarrollar más estudios de investigación sobre farmacorresistencia para así poder mejorar el tratamiento de los pacientes epilépticos actuales y futuros.
6. BIBLIOGRAFÍA
1. Kwan P, Schachter SC, Brodie MJ. Drug-resistant epilepsy. N Engl J Med 2011;365:919- 26.
2. Megiddo I, Colson A, Chisholm D, Dua T, Nandi A, Laxnimarayan R. Health and economic benefits of public financing of epilepsy treatment in India: An agent‐based simulation model. Epilepsia 2016;57:464-74.
3. Banerjee PN, Filippi D, Allen Hauser W. The descriptive epidemiology of epilepsy-a review. Epilepsy Res 2009;85:31-45.
4. Bell GS, Neligan A, Sander, JW. An unknown quantity—the worldwide prevalence of epilepsy. Epilepsia 2014;55:958-62.
5. García-Ramos R, Pastor AG, Masjuan J, Sánchez C, Gil A. FEEN: Informe sociosanitario FEEN sobre la epilepsia en España. Neurología 2011;26:548-55.
6. Rufo-Campos M, Sancho-Rieger J, Peña P, Masramon X, Rejas-Gutierrez J, and colaboradores del estudio LINCE. Pautas terapéuticas en el paciente con epilepsia farmacorresistente en consultas ambulatorias de neurología y epilepsia en España. Rev Neurol 2008;47:517-24.
7. Stephen LJ, Forsyth M, Kelly K, Brodie MJ. Antiepileptic drug combinations–have newer agents altered clinical outcomes? Epilepsy Res 2012;98:194-8.
8. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010;51:1069-77.
9. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:522-30.
10. Brodie MJ, Barry SJE, Bamagous GA, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology 2012;78:1548-54.
11. Corella D, Ordovas JM. Conceptos básicos en biología molecular relacionados con la genética y la epigenética. Rev Esp Cardiol 2017;70:744-53.
12. Golan DE. Principios de farmacología: bases fisiopatológicas del tratamiento farmacológico. 2017. EJ Armstrong & AW Armstrong (Eds.). Lippincott Williams & Wilkins. Philadelphia (EEUU).
13. Alvarez C. Farmacogenética y Farmacorresistencia en la práctica neuropediátrica. Rev Chil 2012;12:118-23.
14. Potschka H, Brodie MJ. Pharmacoresistance. Handb Clin Neurol 2012;108:741-57.
15. Balestrini S, Sisodiya SM. Pharmacogenomics in epilepsy. Neurosci Lett 2018;667:27-39.
16. Shu-xia Li, Yun-yong Liu, Quan-bao Wang. ABCB1 Gene C3435T polymorphism and drug resistance in epilepsy: evidence based on 8604 subjects. Med Sci Monit Basic Res 2015;21:861-8.
17. Parker D, Sanders EJ, Burghardt KJ. Pharmacogenetics of antiepileptic drugs: a brief review. Mental Health Clinician 2016;6:28-34.
18. López-García MA, Feria-Romero IA, Serrano H, Rayo-Mares D, Fagiolino P, Vázquez, M, et al. Influence of genetic variants of CYP2D6, CYP2C9, CYP2C19 and CYP3A4 on antiepileptic drug metabolism in pediatric patients with refractory epilepsy. J Pharmacol Rep 2017;69:504-11.
19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.
20. Walker LE, Mirza N, Yip VL, Marson AG, Pirmohamed M. Personalized medicine approaches in epilepsy. J Intern Med 2015;277:218-34.
7. ENLACES DE INTERÉS:
Organización Mundial de la Salud. (OMS). http://www.who.int/mediacentre/es
International League Against Epilepsy (ILAE). http://www.ilae.org/
Pharmacogenomics knowledge Base. http://www.pharmagkb.org/
Epilepsy Pharmacogenomics: delivering biomarkers for clinical use (EpiPGX). http://www.epipgx.eu/
Canadian Pharmacogenomics Network for Drug Safety (CPNDS). http://cpnds.ubc.ca/